March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part I. Introduction
Chapter 2. Delocalized Chemical Bonding
2.K. Aromatic Systems with Electron Numbers Other than Six
The special stability of benzene is well recognized, and this stability is also associated with rings that are similar, but of different sizes, (e.g., cyclobutadiene (77), cyclooctatetraene (78), cyclodecapentaene (79)212,
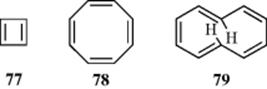
and so on. The general name annulene is given to these compounds,213 benzene being [6]annulene, and 77–79 being called, respectively, [4], [8], and [10]annulene.214 By a naïve consideration of resonance forms, these annulenes and higher ones should be as aromatic as benzene. Yet they proved remarkably elusive. The ubiquitous benzene ring is found in thousands of natural products, in coal and petroleum, and is formed by strong treatment of many noncyclic compounds. None of the other annulene ring systems has ever been found in nature and, except for cyclooctatetraene, their synthesis is not simple. Obviously, there is something special about the number six in a cyclic system of electrons.
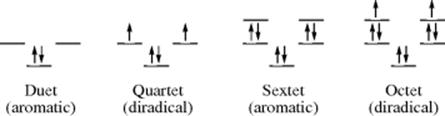
Hückel's rule, based on MO calculations,215 predicts that electron rings will constitute an aromatic system only if the number of electrons in the ring is of the form 4n + 2, where n is zero or any position integer. Systems that contain 4n electrons are predicted to be nonaromatic. The rule predicts that rings of 2, 6, 10, 14, and so on, electrons will be aromatic, while rings of 4, 8, 12, and so on, will not be. This is actually a consequence of Hund's rule. The first pair of electrons in an annulene goes into the π orbital of lowest energy. After that the bonding orbitals are degenerate and occur in pairs of equal energy. When there is a total of four electrons, Hund's rule predicts that two will be in the lowest orbital, but the other two will be unpaired, so that the system will exist as a diradical rather than as two pairs. The degeneracy can be removed if the molecule is distorted from maximum molecular symmetry to a structure of lesser symmetry. For example, if 77 assumes a rectangular rather than a square shape, one of the previously degenerate orbitals has a lower energy than the other and will be occupied by two electrons. In this case, of course, the double bonds are essentially separate and the molecule is still not aromatic. Distortions of symmetry can also occur when one or more carbons are replaced by heteroatoms or in other ways.216 The enthalpy of formation of cyclobutadiene was reported by Kass and co-workers.217 There is a brief discussion of the importance of cyclobutadiene with respect to antiaromaticity.218 A word of caution is in order for MO calculations in these systems. It is known that ab initio computations on benzene at electron-correlated MP2, MP3, CISD, and CCSD levels using a number of popular basis sets219 give anomalous, nonplanar equilibrium structures. 220 The origin of these anomalies has been addressed.220
In the following sections, systems with various numbers of electrons are discussed. Any probe of aromaticity must include (1) the presence of a diamagnetic ring current; (2) equal or approximately equal bond distances, except when the symmetry of the system is disturbed by a heteroatom or in some other way; (3) planarity; (4) chemical stability; (5) the ability to undergo aromatic substitution.
2.K.i Systems of Two Electrons221
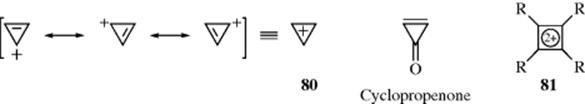
Obviously, there can be no ring of two carbon atoms (a double bond may be regarded as a degenerate case). However, by analogy to the tropylium ion, a three-membered ring with a double bond and a positive charge on the third atom (the cyclopropenyl cation) is a 4n + 2 system and expected to show aromaticity. Unsubstituted 80 has been prepared,222 as well as several derivatives, (e.g., the trichloro, diphenyl, and dipropyl derivatives), and they are stable despite bond angles of only 60°. Tripropylcyclopropenyl,223 tricyclopropylcyclopropenyl,224 chlorodipropylcyclopropenyl,225 and chloro-bis-dialkylaminocyclopropenyl226 cations are among the most stable carbocations known, being stable even in water solution. The tri-tert-butylcyclopropenyl cation is also very stable.227 In addition, cyclopropenone and several of its derivatives are stable compounds,228 in accord with the corresponding stability of the tropones.229 The ring system 80 is nonalternant and the corresponding radical and anion, which do not have an aromatic duet, have electrons in antibonding orbitals, so that their energies are much higher. As with 58 and 62, the equivalence of the three carbon atoms in the triphenylcyclopropenyl cation has been demonstrated by 14C labeling experiments.230 The interesting dications 81 (R = Me or Ph) have been prepared,231 and they too should represent aromatic systems of two electrons.232
2.K.ii. Systems of Four Electrons: Antiaromaticity
The most obvious compound in which to look for a closed loop of four electrons is cyclobutadiene (77).233 Hückel's rule predicts no aromatic character since 4 is not a number generated from 4n + 2. There is a long history of attempts to prepare this compound and its simple derivatives, and those experiments fully bear out Hückel's prediction. Cyclobutadienes display none of the characteristics that would lead us to call them aromatic, and there is evidence that a closed loop of four electrons is actually antiaromatic.234 If such compounds simply lacked aromaticity, we would expect them to be about as stable as similar nonaromatic compounds, but both theory and experiment show that they are much less stable.235 An antiaromatic compound may be defined as a compound that is destabilized by a closed loop of electrons.
Cyclobutadiene was first prepared by Pettit and co-workers.236 It is now clear that 77 and its simple derivatives are extremely unstable compounds with very short lifetimes (they dimerize by a Diels–Alder reaction; see 15–60) unless they are stabilized in some fashion, either at ordinary temperatures embedded in the cavity of a hemicarcerand237 (see the structure of a carcerand in Sec. 3.C.iii), or in matrices at very low temperatures (generally < 35 K). In either of these cases, the cyclobutadiene molecules are forced to remain apart from each other, and other molecules cannot get in. The structures of 77 and some of its derivatives have been studied a number of times using the low-temperature matrix technique.238 The ground-state structure of 77 is a rectangular diene (not a diradical), as shown by the (Ir) spectra of 77 and deuterated 77 trapped in matrices,239 as well as by a photoelectron spectrum.240Molecular orbital calculations agree.241 The same conclusion was also reached in an elegant experiment in which 1,2-dideuterocyclobutadiene was generated. If 77 is a rectangular diene, the dideutero compound should exist as two isomers, as shown.
The compound was generated (as an intermediate that was not isolated) and two isomers were indeed found.242 The cyclobutadiene molecule is not static, even in the matrices. There are two forms (77a and 77b) that rapidly interconvert.243 Note that there is experimental evidence that the aromatic and antiaromatic characters of neutral and dianionic systems are measurably increased via deuteration.244
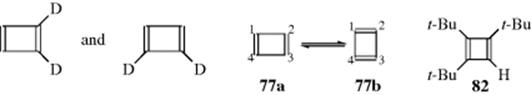
There are some simple cyclobutadienes that are stable at room temperature for varying periods of time. These either have bulky substituents or carry certain other stabilizing substituents, such as seen in tri-tert-butylcyclobutadiene (83).245 Such compounds are relatively stable because dimerization is sterically hindered. Examination of the NMR spectrum of 82 showed that the ring proton (δ = 5.38) was shifted upfield, compared with the position expected for a nonaromatic proton, (e.g., cyclopentadiene). As will be seen in Section. 2.K.vi, this indicates that the compound is antiaromatic.
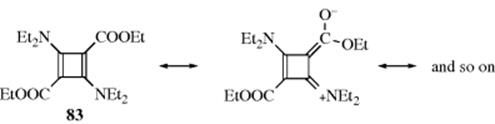
The other type of stable cyclobutadiene has two electron-donating and two electron-withdrawing groups,246 and is stable in the absence of water.247 An example is 83. The stability of these compounds is generally attributed to the resonance shown, a type of resonance stabilization called the push–pull or captodative effect,248 although it has been concluded from a PES that second-order bond fixation is more important.249 An X-ray crystallographic study of 83has shown250 the ring to be a distorted square with bond lengths of 1.46 Å and angles of 87° and 93°.
It is clear that simple cyclobutadienes, which could easily adopt a square planar shape if that would result in aromatic stabilization, do not in fact do so and are not aromatic. The high reactivity of these compounds is not caused merely by steric strain, since the strain should be no greater than that of simple cyclopropenes, which are known compounds. It is probably caused by antiaromaticity.251
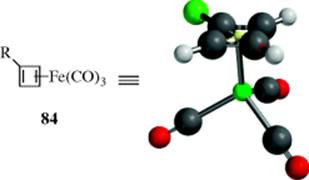
The cyclobutadiene system can be stabilized as a η4-complex with metals,252 as with the iron complex 84 (see Chap 3), but in these cases electron density is withdrawn from the ring by the metal and there is no aromatic quartet. In fact, these cyclobutadiene–metal complexes can be looked upon as systems containing an aromatic duet. The ring is square planar,253 the compounds undergo aromatic substitution,254 and NMR spectra of monosubstituted derivatives show that the C-2 and C-4 protons are equivalent.229
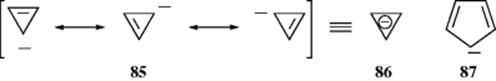
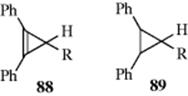
Other systems that have been studied as possible aromatic or antiaromatic four-electron systems include the cyclopropenyl anion (86) and the cyclopentadienyl cation (87).255 With respect to 86, HMO theory predicts that an unconjugated 85 (i.e., a single canonical form) is more stable than a conjugated 86,256 so that 85 would actually lose stability by forming a closed loop of four electrons. The HMO theory is supported by experiment. Among other evidence, it has been shown that 88 (R = COPh) loses its proton in hydrogen-exchange reactions ~ 6000 times more slowly than 89 (R = COPh).257 Where R = CN, the ratio is ~ 10,000.258 This indicates that 88 are much more reluctant to form carbanions (which would have to be cyclopropenyl carbanions) than 89, which form ordinary carbanions. Thus the carbanions of 88 are less stable than corresponding ordinary carbanions. Although derivatives of cyclopropenyl anion have been prepared as fleeting intermediates (as in the exchange reactions mentioned above), all attempts to prepare the ion or any of its derivatives as relatively stable species have so far met with failure.259
In the case of 87, the ion has been prepared and shown to be a diradical in the ground state,260 as predicted by the discussion in Section 2.K.ii.261 Evidence that 87 is not only nonaromatic, but is antiaromatic comes from studies on 90 and 92.262 When 90 is treated with silver perchlorate in propionic acid, the molecule is rapidly solvolyzed (a reaction in which the intermediate 91 is formed; see Chapters 5 and 10). Under the same conditions, 92 undergoes no solvolysis at all; that is, 87 does not form. If 87 were merely nonaromatic, it should be about as stable as 91 (which of course has no resonance stabilization at all). The fact that it is so much more reluctant to form indicates that 87 is much less stable than 91. Note that under certain conditions, 91 can be generated solvolytically.263
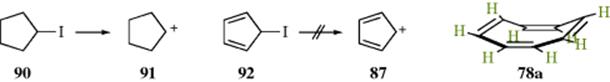
The fact that 86 and 87 are not aromatic while the cyclopropenyl cation (80) and the cyclopentadienyl anion (58) is strong evidence for Hückel's rule since simple resonance theory predicts no difference between 86 and 80 or 87and 58 (the same number of equivalent canonical forms can be drawn for 86 as for 80 and for 87 as for 58).
2.K.iii. Systems of Eight Electrons
Cyclooctatetraene264 ([8]annulene, 78a) is not planar, but tub shaped,265 so it is neither aromatic nor antiaromatic, since both these conditions require overlap of parallel p orbitals. The reason for the lack of planarity is that a regular octagon has angles of 135°, while sp2 angles are most stable at 120°. To avoid the strain, the molecule assumes a nonplanar shape, in which orbital overlap is greatly diminished.266 Single- and double-bond distances in 78 are, respectively, 1.46 and 1.33 Å, which is expected for a compound made up of four individual double bonds.265 The Jahn–Teller effect has been invoked to explain the instability of such antiaromatic compounds.267 The Jahn-Teller effect arises from molecular distortions due to an electronically degenerate ground state.268
The reactivity is also what would be expected for a linear polyene. Reactive intermediates can be formed in solution. Dehydrohalogenation of bromocyclooctatetraene at −100 °C has been reported, for example, and trapping by immediate electron transfer gave a stable solution of the [8]annulyne anion radical.269
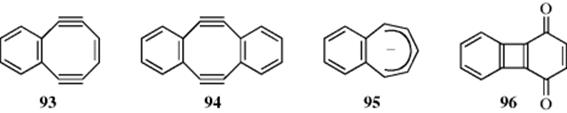
The cyclooctadiendiynes 93 and 94 are planar conjugated eight-electron systems (the four extra triple-bond electrons do not participate), which NMR evidence show to be antiaromatic.270 There is evidence that part of the reason for the lack of planarity in 78 itself is that a planar molecule would have to be antiaromatic.271 The cycloheptatrienyl anion (61) also has eight electrons, but does not behave like an aromatic system.167 The bond lengths for a series of molecules containing the cycloheptatrienide anion have recently been published.272 The NMR spectrum of the benzocycloheptatrienyl anion (95) shows that, like 82, 93, and 94, this compound is antiaromatic.273 A new anti-aromatic compound 1,4-biphenylene quinone (96) was prepared, but it rapidly dimerizes due to instability.274
2.K.iv. Systems of Ten Electrons275
There are three possible geometrical isomers of [10]annulene: the all-cis (97), the mono-trans (98), and the cis-trans-cis-cis-trans (79). If Hückel's rule applies, they should be planar. But it is far from obvious that the
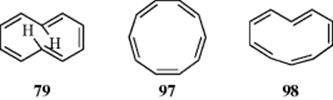
molecules would adopt a planar shape, since they must overcome considerable strain to do so. For a regular decagon (97) the angles would have to be 144°, considerably larger than the 120° required for sp2 angles. Some of this strain would also be present in 98, but this kind of strain is eliminated in 79 since all the angles are 120°. However, it was pointed out by Mislow276 that the hydrogen atoms in the 1 and 6 positions should interfere with each other and force the molecule out of planarity. Such configurational changes are not necessarily without cost energetically. It has been determined that configurational changes in [14]annulene, for example. requires Möbius antiaromatic bond shifting.277
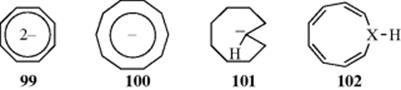
Compounds 97 and 98 have been prepared278 as crystalline solids at −80 °C. The NMR spectra show that all the hydrogen atoms lie in the alkene region, and it was concluded that neither compound is aromatic. Calculations on 98suggest that it may indeed be aromatic, although the other isomers are not.279 It is known that the Hartree–Fock (HF) method incorrectly favors bond-length-alternating structures for [10]annulene, and aromatic structures are incorrectly favored by density functional theory. Improved calculations predict that the twist conformation is lowest in energy, and the naphthalene-like and heart-shaped conformations lie higher than the twist by 1.40 and 4.24 kcal mol−1(5.86 and 17.75 kJ mol−1), respectively.280 Analysis of 13C and 1H NMR spectra suggest that neither is planar. However, the preparation of several compounds that have large angles, but that are definitely planar 10-electron aromatic systems, clearly demonstrate that the angle strain is not insurmountable. Among these are the dianion 99, the anions 100 and 101, and the azonine 102.281 Compound 99282 has angles of ~ − 135°, while 100283 and 101284have angles of ~ 140°, which are not very far from 144°. The inner proton in 101285 which is the mono-trans isomer of the all-cis 100 is found far upfield in the NMR (−3.5 δ). For 97 and 98, the cost in strain energy to achieve planarity apparently outweighs the extra stability that would come from an aromatic ring. Further emphasizing the delicate balance between these factors, it is known that the oxygen analogue of 102 (X = O, oxonin) and the N-carbethoxy derivative of 102 (X = CH) are nonaromatic and nonplanar, while 102 (X = N) is aromatic and planar.286 Other azaannulenes are known, including Vogel's 2,7-methanoazaannulene,287 as well as 3,8-methanoaza[10]annulene,288 and the alkoxy derivative of both.289 Calculations for aza[10]annulene concluded that the best olefinic twist isomer is 2.1 kcal mol−1 (8.8 kJ mol−1) more stable than the aromatic form,290 and is probably the more stable form.
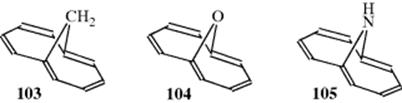
So far, 79 from above has not been prepared despite many attempts. However, there are various ways of avoiding the interference between the two inner protons. The approach that has been most successful involves bridging the 1 and 6 positions.291 Thus, 1,6-methano[10]annulene (103)292 and its oxygen and nitrogen analoges 104293 and 105294 have been prepared, and they are stable compounds, are diatropic, and undergo aromatic substitution.295 For example, the perimeter protons of 103 are found at 6.9–7.3 δ, while the bridge protons are at −0.5 δ. The crystal structure of 103 shows that the perimeter is nonplanar, but the bond distances are in the range 1.37–1.42 Å.296 It has therefore been amply demonstrated that a closed loop of 10 electrons is an aromatic system, although some molecules that could conceivably have such a system are too distorted from planarity to be aromatic. A small distortion from planarity (as in 103) does not prevent aromaticity, at least in part because the σ orbitals so distort themselves as to maximize the favorable (parallel) overlap of p orbitals to form the aromatic 10-electron loop.297
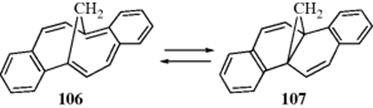
In 106, where 103 is fused to two benzene rings in such a way that no canonical form can be written in which both benzene rings have six electrons, the aromaticity is diminished by annellation, as shown by the fact that the molecule rapidly converts to the more stable 107, in which both benzene rings can be fully aromatic298 (this is similar to the cycloheptatriene–norcaradiene conversions discussed in 18–32).
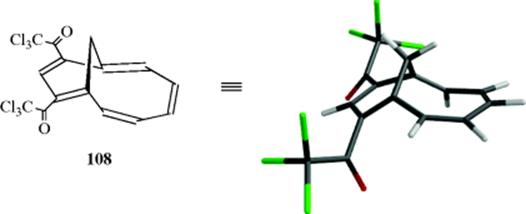
Molecules can sustain significant distortion from planarity and retain their aromatic character. 1,3-Bis(trichloroacetyl)homoazulene (108) qualifies as aromatic using the geometric criterion that there is only a small average deviation from the C–C bond length in the [10]annulene perimeter.299 The X-ray crystal structure shows that the 1,5-bridge distorts the [10]annulene π-system away from planarity (see the 3D model) with torsion angles as large as 42.2° at the bridgehead position, but 108 does not lose its aromaticity.
2.K.v. Systems of More Than Ten Electrons: 4n + 2 Electrons300
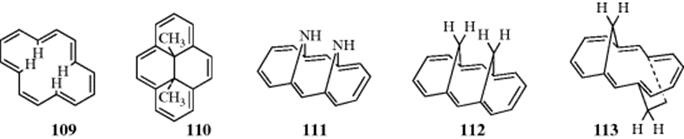
Extrapolating from the discussion of [10]annulene, larger 4n + 2 systems are expected to be aromatic if they are planar. Mislow276 predicted that [14]annulene (109) would possess the same type of interference as 79, although to a lesser degree. This is borne out by experiment. Compound 109 is aromatic (it is diatropic; inner protons at 0.00 δ, outer protons at 7.6 δ),301 but is highly reactive and is completely destroyed by light and air in 1 day. X-ray analysis shows that although there are no alternating single and double bonds (the molecule is not planar).302 A number of stable bridged [14]annulenes have been prepared303 [e.g., trans-15,16-dimethyldihydropyrene (110),304syn-1,6:8,13-diimino[14]annulene (111),305 and syn- and anti-1,6:8,13-bismethano[14]annulene (112 and 113)].306 The dihydropyrene (110, and its diethyl and dipropyl homologues) is undoubtedly aromatic: The π perimeter is approximately planar,307 the bond distances are all 1.39–1.40 Å, and the molecule undergoes aromatic substitution304 as well as being diatropic.308 The outer protons are found at 8.14–8.67 δ, while the CH3 protons are at −4.25 δ. Other nonplanar aromatic dihydropyrenes are known.309 Annulenes 111 and 112 are also diatropic,310 although X-ray crystallography indicates that the π periphery in 111 is not quite planar.311 In 113, the geometry of the molecule greatly reduces the overlap of the p orbitals at the bridgehead positions with adjacent p orbitals, and it is definitely not aromatic,312 as shown by NMR spectra306 and X-ray crystallography, from which bond distances of 1.33–1.36 Å for the double bonds and 1.44–1.49 Å for the single bonds have been obtained.313 In contrast, all the bond distances in 111 are ~ 1.38–1.40 Å.311
Another way of eliminating the hydrogen interferences of [14]annulene is to introduce one or more triple bonds into the system, as in dehydro[14]annulene (114).314 All five known dehydro[14]annulenes are diatropic, and 87 can be nitrated or sulfonated.315 The extra electrons of the triple bond do not form part of the
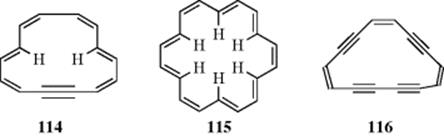
aromatic system, but it simply exists as a localized bond. There has been a debate concerning the extent of delocalization in dehydrobenzoannulenes,316 but there is evidence for a weak, but discernible ring current.3173,4,7,8,9,10,13,14-Octahydro[14]annulene (116) has been prepared, for example, and the evidence supported its aromaticity.318 This study suggested that increasing benzoannelation of the parent 116 led to a step-down in aromaticity, a result of competing ring currents in the annulenic system. Note that [12]annulyne has been prepared.319
[18]Annulene (115) is diatropic:320 the 12 outer protons are found at ~ δ = 9 and the 6 inner protons at ~ δ = −3. X-ray crystallography321 shows that it is nearly planar, so that interference of the inner hydrogen atoms is not important in annulenes this large. Compound 115 is reasonably stable, being distillable at reduced pressures, and undergoes aromatic substitutions322 (Chapter 11). The C–C bond distances are not equal, but they do not alternate. There are 12 inner bonds of ~1.38 Å and 6 outer bonds of ~1.42 Å.321 Compound 115 has been estimated to have a resonance energy of ~37 kcal mol−1 (155 kJ mol−1), similar to that of benzene.323
The known bridged [18]annulenes are also diatropic,324 as are most of the known dehydro[18]annulenes.325 The dianions of open and bridged [16]annulenes326 are also 18-electron aromatic systems,327 and there are dibenzo[18]annulenes.328
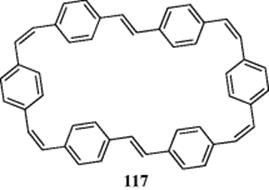
[22]Annulene329 and dehydro[22]annulene330 are also diatropic. A dehydrobenzo[22]annulene has been prepared that has eight C![]() C units, is planar, and possesses a weak induced ring current.331 In the latter compound, there are 13 outer protons at 6.25–8.45 δ and 7 inner protons at 0.70–3.45 δ. Some aromatic bridged [22]annulenes are known.332 [26]Annulene has not yet been prepared, but several dehydro[26]annulenes are aromatic.333 Furthermore, the dianion of 1,3,7,9,13,15,19,21-octadehydro[24]annulene is another 26-electron system that is aromatic.334 Ojima and et al.335 prepared bridged dehydro derivatives of [26], [30], and [34]annulenes. All are diatropic. The same workers prepared a bridged tetradehydro[38]annulene,335 which showed no ring current. On the other hand, the dianion of the cyclophane, (117) also has 38 perimeter electrons, and this species is diatropic.336
C units, is planar, and possesses a weak induced ring current.331 In the latter compound, there are 13 outer protons at 6.25–8.45 δ and 7 inner protons at 0.70–3.45 δ. Some aromatic bridged [22]annulenes are known.332 [26]Annulene has not yet been prepared, but several dehydro[26]annulenes are aromatic.333 Furthermore, the dianion of 1,3,7,9,13,15,19,21-octadehydro[24]annulene is another 26-electron system that is aromatic.334 Ojima and et al.335 prepared bridged dehydro derivatives of [26], [30], and [34]annulenes. All are diatropic. The same workers prepared a bridged tetradehydro[38]annulene,335 which showed no ring current. On the other hand, the dianion of the cyclophane, (117) also has 38 perimeter electrons, and this species is diatropic.336
There is now no doubt that 4n + 2 systems are aromatic if they can be planar, although 97 and 113 among others, demonstrate that not all such systems are in fact planar enough for aromaticity. Both 109 and 111 prove that absolute planarity is not required for aromaticity, but that aromaticity decreases with decreasing planarity.
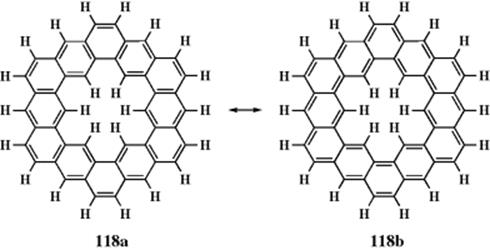
The ![]() NMR spectrum of 118 (called kekulene) showed that in a case where electrons can form either aromatic sextets or larger systems, the sextets are preferred.337 There was initial speculation that kekulene might be superaromatic, that is, it would show enhanced aromatic stabilization. Calculations suggest that there is no enhanced stabilization.338 The 48 π electrons of 118 might, in theory, prefer structure 118a, where each ring is a fused benzene ring, or 118b, which has a [30]annulene on the outside and an [18]annulene on the inside. The
NMR spectrum of 118 (called kekulene) showed that in a case where electrons can form either aromatic sextets or larger systems, the sextets are preferred.337 There was initial speculation that kekulene might be superaromatic, that is, it would show enhanced aromatic stabilization. Calculations suggest that there is no enhanced stabilization.338 The 48 π electrons of 118 might, in theory, prefer structure 118a, where each ring is a fused benzene ring, or 118b, which has a [30]annulene on the outside and an [18]annulene on the inside. The ![]() NMR spectrum of this compound shows three peaks at δ = 7.94, 8.37, and 10.45 in a ratio of 2:1:1. Examination of the structure shows that 118 contains three groups of protons. The peak at 7.94 δ is attributed to the 12 ortho protons and the peak at 8.37 δ to the six external para protons. The remaining peak comes from the six inner protons. If the molecule preferred 118b, this peak should be upfield, probably with a negative δ, as in the case of 115. The fact that this peak is far downfield indicates that the electrons prefer to be in benzenoid rings. Note that in the case of the dianion of 117, the opposite situation prevails. In this ion, the 38-electron system is preferred even though 24 of these must come from the six benzene rings, which therefore cannot have aromatic sextets.
NMR spectrum of this compound shows three peaks at δ = 7.94, 8.37, and 10.45 in a ratio of 2:1:1. Examination of the structure shows that 118 contains three groups of protons. The peak at 7.94 δ is attributed to the 12 ortho protons and the peak at 8.37 δ to the six external para protons. The remaining peak comes from the six inner protons. If the molecule preferred 118b, this peak should be upfield, probably with a negative δ, as in the case of 115. The fact that this peak is far downfield indicates that the electrons prefer to be in benzenoid rings. Note that in the case of the dianion of 117, the opposite situation prevails. In this ion, the 38-electron system is preferred even though 24 of these must come from the six benzene rings, which therefore cannot have aromatic sextets.
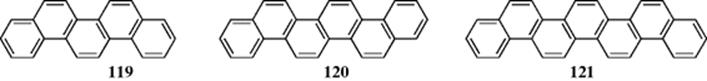
Phenacenes are a family of “graphite ribbons,” where benzene rings are fused together in an alternating pattern. Phenanthrene is the simplest member of this family and other members include the 22 electron system picene (119), the 26 electron system fulminene (120) and the larger member of this family, the 30 electron [7]phenancene, with seven rings (121).339 In the series benzene to heptacene, reactivity increases although acene resonance energies per π electron are nearly constant. The inner rings of the “acenes” are more reactive, and calculations showed that those rings are more aromatic than the outer rings, and are even more aromatic than benzene itself.340 N-Heteroacenes are also known.341
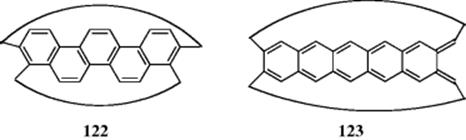
A super-ring molecule is formed by rolling a polyacene molecule into one ring with one edge benzene ring folding into the other. These are called cyclopolyacenes or cyclacenes.342 Although the zigzag cyclohexacenes (122) are highly aromatic (this example is a 22 electron system), the linear cyclohexacenes (e.g., the 24 electron 123) are much less aromatic.343 It is possible to deform an acene by attaching bulky substituents to its periphery by single covalent bonds. The presence of these substituents, which often leads to twisting of torsion angles, is usually easier than distortion of bond angles or C-C bond lengths. Such compounds are called twisted acenes.344
2.K.vi. Systems of More Than 10 Electrons: 4n Electrons249
As seen in Section 2.K.ii, these systems are expected to be not only nonaromatic, but also antiaromatic. The [12]annulene (124) has been prepared.345 In solution, 124 exhibits rapid conformational mobility (as do many other annulenes),346 and above −150°C in this particular case, all protons are magnetically equivalent. However, at −170°C the mobility is greatly slowed and the three inner protons are found at ~ 8 δ while the nine
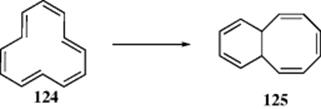
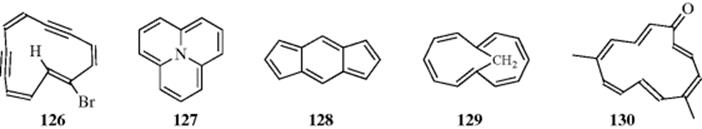
outer protons are at ~ 6 δ. Interaction of the ‘internal’ hydrogen atoms in annulene (124) leads to nonplanarity. Above −50°C, 124 is unstable and rearranges to 125. Several bridged and dehydro[12]annulenes are known, for example, 5-bromo-1,9-didehydro[12]annulene (126),347 cycl[3.3.3]azine (127),348s-indacene (128),349 and 1,7-methano[12]annulene (129).350s-Indacene is a planar, conjugated system perturbed by two cross links, and studies showed that the low-energy structure has localized double bonds. In these compounds, both hydrogen interference and conformational mobility are prevented. In 127–129, the bridge prevents conformational changes, while in 126the bromine atom is too large to be found inside the ring. The NMR spectra show that all four compounds are paratropic, the inner proton of 126 being found at 16.4 δ. The dication of 112351 and the dianion of 103352 are also 12-electron paratropic species. An interesting 12-electron [13]annulenone has recently been reported, 5,10-dimethyl[13]annulenone (130). This annulenone is the first monocyclic annulene larger than tropane,353 and a linearly–fused benzodehydro[12]annulene system has been reported.354
The results for [16]annulene are similar. The compound was synthesized in two different ways,355 both of which gave 131, which in solution is in equilibrium with 132. Above −50 °C there is conformational mobility, resulting in the magnetic equivalence of all protons, but at −130 °C the compound is clearly paratropic: There are 4 protons at 10.56 δ and 12 at 5.35 δ. In the solid state, where the compound exists entirely as 131, X-ray crystallography356shows that the molecules are nonplanar with almost complete bond alternation: The single bonds are 1.44–1.47 Å and the double bonds are 1.31–1.35 Å. A number of dehydro and bridged [16]annulenes are also paratropic,357 as are [20]annulene,358 and [24]annulene.359 However, a bridged tetradehydro[32]annulene was atropic.333
Both pyracyclene (133),360 which because of strain is stable only in solution, and dipleiadiene (134)361 are paratropic, as shown by NMR spectra. These molecules might have been expected to behave like naphthalenes with outer bridges, but the outer π frameworks (12 and 16 electrons, respectively) constitute antiaromatic systems with an extra central double bond. With respect to 133, the 4n + 2 rule predicts pyracylene to be “aromatic” if it is regarded as a 10-π-electron naphthalene unit connected to two 2-π-electron etheno systems, but “antiaromatic” if it is viewed as a 12-π-electron cyclododecahexaene periphery perturbed by an internal cross-linked etheno unit.362 Recent studies have concluded on energetic grounds that 133 is a “borderline” case, in terms of aromaticity–antiaromaticity character.360 Dipleiadiene appears to be antiaromatic.361
The fact that many 4n systems are paratropic even though they may be nonplanar and have unequal bond distances indicates that if planarity were enforced, the ring currents might be even greater. The NMR spectrum of the dianion of 110363 (and its diethyl and dipropyl homologues)364 effectively illustrate this point. Recall that in 110, the outer protons were found at 8.14–8.67 δ with the methyl protons at −4.25 δ. For the dianion, however, which is forced to have approximately the same planar geometry, but now has 16 electrons, the outer protons are shifted to about −3 δ while the methyl protons are found at ~ 21 δ, a shift of ~ 25 δ! A converse shift was made when [16]annulenes that were antiaromatic were converted to 18-electron dianions that were aromatic.282 In these cases, the changes in NMR chemical shifts were almost as dramatic. Heat-of-combustion measures also show that [16]annulene is much less stable than its dianion.365 It has also been reported that the fluorenyl cation shows substantial destabilization, suggesting that it is an antiaromatic species.366
It seems clear that 4n systems will be at a maximum where a molecule is constrained to be planar (as in 86 or the dianion of 110) but, where possible, the molecule will distort itself from planarity and avoid equal bond distances in order to reduce. In some cases (e.g., cyclooctatraene), the distortion and bond alternation are great enough to be completely avoided. In other cases, (e.g., 124 or 131), it is apparently not possible for the molecules to avoid at least some p orbital overlap. Such molecules show evidence of paramagnetic ring currents, although the degree of is not as great as in molecules (e.g., 86 or the dianion of 110).
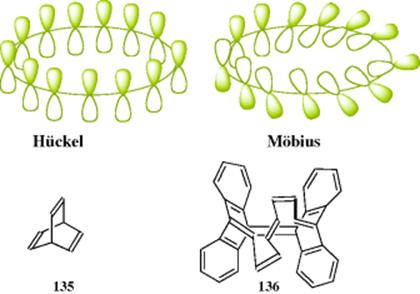
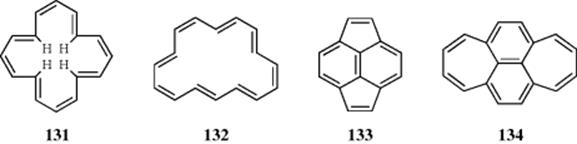
The concept of “Möbius aromaticity” was conceived by Helbronner in 1964,367 when he suggested that large cyclic [4n]annulenes might be stabilized if the π orbitals were twisted gradually around a Möbius strip. This concept is illustrated by the diagrams labeled Hückel, which is a destabilized [4n] system, in contrast to the Möbius model, which is a stabilized [4n] system.368 Zimmerman generalized this idea and applied the “Hückel–Möbius concept” to the analysis of ground-state systems [e.g., barrelene (135)].369 In 1998, a computational reinterpretation of existing experimental evidence for (CH)9+ as a Möbius aromatic cyclic annulene with 4n π-electrons was reported.370 This reversal of [4n]annulene antiaromaticity has been demonstrated by stacking rings into a superphane.371 A superphane is a sixfold bridged cyclophane with all arene positions in the corresponding dimer taken up by ethylene spacers.372 A recent computational study predicted several Möbius local minima for [12], [16], and [20]annulenes.373 A twisted [16]annulene has been prepared and calculations suggested it should show Möbius aromaticity.374 High-performance liquid chromatography (HPLC) separation of isomers gave 136, and the authors concluded it is Möbius aromatic. The synthesis and study of molecules that demonstrate Möbius aromaticity continues to be an area of interest.375