March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part I. Introduction
Chapter 5. Carbocations, Carbanions, Free Radicals, Carbenes, and Nitrenes
5.C. Free Radicals
5.C.i. Stability and Structure211
A free radical (usually just called a radical) may be defined as a species that contains one or more unpaired electrons. Note that this definition includes certain stable inorganic molecules (e.g., NO and NO2), as well as many individual atoms (e.g., Na and Cl). As with carbocations and carbanions, simple alkyl radicals are very reactive and are usually transient species. For the most part, their lifetimes are extremely short in solution, but they can be kept frozen for relatively long periods of time within the crystal lattices of other molecules.212 There are, however, many stable radicals,213 some of which will be noted below. Many spectral214 measurements have been made on radicals trapped in this manner. Even under these conditions the methyl radical decomposes with a half-life of 10–15 min in a methanol lattice at 77 K.215 Since the lifetime of a radical depends not only on its inherent stability, but also on the conditions under which it is generated, the terms persistent and stable are usually used for the different senses. A stable radical is inherently stable; a persistent radical has a relatively long lifetime under the conditions at which it is generated, although it may not be very stable.
Radicals can be characterized by several techniques (e.g., mass spectrometry216 or the characterization of alkoxycarbonyl radicals by Step–Scan Time-Resolved Infrared Spectroscopy).217 Another technique makes use of the magnetic moment that is associated with the spin of an electron, which can be expressed by a quantum number of ![]() or
or ![]() . According to the Pauli principle, any two electrons occupying the same orbital must have opposite spins, so the total magnetic moment is zero for any species in which all the electrons are paired. In radicals, however, one or more electrons are unpaired, so there is a net magnetic moment and the species is paramagnetic. Radicals can therefore be detected by magnetic-susceptibility measurements, but for this technique a relatively high concentration of radicals is required.
. According to the Pauli principle, any two electrons occupying the same orbital must have opposite spins, so the total magnetic moment is zero for any species in which all the electrons are paired. In radicals, however, one or more electrons are unpaired, so there is a net magnetic moment and the species is paramagnetic. Radicals can therefore be detected by magnetic-susceptibility measurements, but for this technique a relatively high concentration of radicals is required.
A much more important technique is electron spin resonance (ESR), also called electron paramagnetic resonance (EPR).218 The principle of ESR is similar to that of NMR, except that electron spin is involved rather than nuclear spin. The two electron spin states (![]() and
and ![]() ) are ordinarily of equal energy, but in a magnetic field the energies are different. As in NMR, a strong external field is applied and electrons are caused to flip from the lower state to the higher by the application of an appropriate radio frequency signal. Inasmuch as two electrons paired in one orbital must have opposite spins that cancel, an ESR spectrum arises only from species that have one or more unpaired electrons (i.e., free radicals).
) are ordinarily of equal energy, but in a magnetic field the energies are different. As in NMR, a strong external field is applied and electrons are caused to flip from the lower state to the higher by the application of an appropriate radio frequency signal. Inasmuch as two electrons paired in one orbital must have opposite spins that cancel, an ESR spectrum arises only from species that have one or more unpaired electrons (i.e., free radicals).
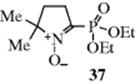
Since only free radicals give an ESR spectrum, the method can be used to detect the presence of radicals and to determine their concentration.219 Furthermore, information concerning the electron distribution (and hence the structure) of free radicals can be obtained from the splitting pattern of the ESR spectrum (ESR peaks are split by nearby protons).220 Fortunately (for the existence of most free radicals is very short), it is not necessary for a radical to be persistent for an ESR spectrum to be obtained. Electron spin resonance spectra have been observed for radicals with lifetimes considerably <1 s. Failure to observe an ESR spectrum does not prove that radicals are not involved, since the concentration may be too low for direct observation. In such cases, the spin-trapping technique can be used.221 In this technique, a compound is added that is able to combine with very reactive radicals to produce more persistent radicals; the new radicals can be observed by ESR. Azulenyl nitrones have been developed as chromotropic spin-trapping agents.222 An important class of spin-trapping compounds are nitroso compounds, which react with radicals to give stable nitroxide radicals:223 RN=O + R′√ → RR′N–O√. An N-oxide spin trap has been developed [37; 2(diethylphosphino)-5,5-dimethyl-1-pyrroline-N-oxide], and upon trapping a reactive free radical, 31P NMR can be used to identify it.224 This technique is effective, and short-lived species (e.g., the oxiranylmethyl radical) have been detected by spin trapping.225 Other molecules have been used to probe the intermediacy of radicals via SET processes. They are called SET probes.226
Because there is an equal probability that a given unpaired electron will have a quantum number of ![]() or
or ![]() , radicals are observed as a single line in an ESR spectrum unless they interact with other electronic or nuclear spins or possess magnetic anisotropy, in which case two or more lines may appear in the spectrum.227
, radicals are observed as a single line in an ESR spectrum unless they interact with other electronic or nuclear spins or possess magnetic anisotropy, in which case two or more lines may appear in the spectrum.227
Another magnetic technique for the detection of free radicals uses an ordinary NMR instrument. It was discovered228 that if an NMR spectrum is taken during the course of a reaction, certain signals might be enhanced, either in a positive or negative direction; others may be reduced. When this type of behavior, called chemically induced dynamic nuclear polarization229 (CIDNP), is found in the NMR spectrum of the product of a reaction, it means that at least a portion of that product was formed via the intermediacy of a free radical.230 For example, the question was raised whether radicals were intermediates in the exchange reaction between ethyl iodide and ethyllithium (Reaction 12-39):
![]()
Curve a in Fig. 5.1231 shows an NMR spectrum taken during the course of the reaction. Curve b is a reference spectrum of ethyl iodide (CH3 protons at δ = 1.85; CH2 protons at δ = 3.2). Note that in curve a some of the ethyl iodide signals are enhanced; others go below the base line (negative enhancement; also called emission). Thus the ethyl iodide formed in the exchange shows CIDNP and so was formed via a free radical intermediate. Chemically induced dynamic nuclear polarization results when protons in a reacting molecule become dynamically coupled to an unpaired electron while traversing the path from reactants to products. Although the presence of CIDNP almost always means that a free radical is involved,232 its absence does not prove that a free radical intermediate is necessarily absent, since reactions involving free radical intermediates can also take place without observable CIDNP. Also, the presence of CIDNP does not prove that all of a product was formed via a free radical intermediate, only that some of it was. Note that dynamic nuclear polarization (DNP) enhances signal intensities in the NMR spectra of solids and liquids. In a contemporary DNP experiment, a diamagnetic sample is doped with a paramagnet and the large polarization of the electron spins is transferred to the nuclei via microwave irradiation of the EPR spectrum.233 Dynamic nuclear polarization has been used to examine biradicals.234
Fig. 5.1231 (a) The NMR spectrum taken during reaction between EtI and EtLi in benzene (the region between 0.5 and 3.5 δ was scanned with an amplitude twice that of the remainder of the spectrum). The signals at 1.0–1.6 δ are due to butane, some of which is also formed in the reaction. (b) Reference spectrum of EtI. [Reprinted with permission from Ward, H.R.; Lawler, R.G.; Cooper, R.A. J. Am. Chem. Soc. 1969, 91, 746. Copyright © 1969 American Chemical Society.]

As with carbocations, the stability order of free radicals is tertiary > secondary > primary, explainable by field effects and hyperconjugation, analogous to that in carbocations (Sec. 5.A.ii).235
With resonance possibilities, the stability of free radicals increases;236 some can be kept indefinitely.237 Benzylic and allylic238 radicals for which canonical forms can be drawn similar to those shown for the corresponding cations (Sec. 5.A.ii) and anions (Sec. 5.B.i, category 1) are more stable than simple alkyl radicals, but still have only a transient existence under ordinary conditions. Note that 2-phenylethyl radicals have been shown to exhibit bridging of the phenyl group.239
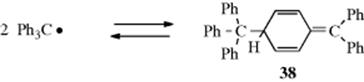
The triphenylmethyl and similar radicals240 are stable enough to exist in solution at room temperature, although they are in equilibrium with a dimeric form. The concentration of triphenylmethyl radical in benzene solution is ~2% at room temperature. For many years, it was assumed that Ph3C√, the first stable free radical known,241 dimerized to hexaphenylethane (Ph3C–CPh3),242 but UV and NMR investigations have shown that the true structure is 38.243Although triphenylmethyl-type radicals are stabilized by resonance:
![]()
steric hindrance to dimerization and not resonance is the major cause of their stability.244 This was demonstrated by the preparation of the radicals 39 and 40.245 These radicals are electronically very similar, but 39, being planar, has much less steric hindrance to dimerization than Ph3C√, while 40, with six groups in ortho positions, has much more. On the other hand, the planarity of 35 means that it has a maximum amount of
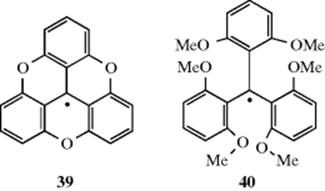
resonance stabilization, while 40 must have much less, since its degree of planarity should be even less than Ph3C√, which itself is propeller shaped and not planar. Thus if resonance is the chief cause of the stability of Ph3C√, 40should dimerize and 39 should not, but if steric hindrance is the major cause, the reverse should happen. It was found233 that 40 gave no evidence of dimerization, even in the solid state, while 39 existed primarily in the dimeric form, which is dissociated to only a small extent in solution.246 This result indicates that steric hindrance to dimerization is the major cause for the stability of triarylmethyl radicals. A similar conclusion was reached in the case of (NC)3C√, which dimerizes readily although it is considerably stabilized by resonance.247 Nevertheless, that resonance is still an important contributing factor to the stability of radicals is shown by the facts that (1) the radical t-Bu(Ph)2C√ dimerizes more than Ph3C√, while p-PhCOC6H4(Ph2)C√ dimerizes less.248 The latter has more canonical forms than Ph3C√, but steric hindrance should be about the same (for attack at one of the two rings). (2) A number of radicals (p-XC6H4)3C√, with X = F, Cl, O2N, CN, and so on, do not dimerize, but are kinetically stable.249 Completely chlorinated triarylmethyl radicals are more stable than the unsubstituted kind, probably for steric reasons, and many are quite inert in solution and in the solid state.250
Allylic radical are relatively stable, and the pentadienyl radical is particularly stable, but (E,E)-, (E,Z)-, and (Z,Z)-stereoisomers can form. It has been calculated that the (Z,Z)-pentadienyl radical is 5.6 kcal mol−1 less stable than the (E,E)-pentadienyl radical.251 Note that vinyl radicals have (E)- and (Z)-forms and the inversion barrier from one to the other increases as the electronegativity of substituents increase.252 Conjugated propargylic radicals are calculated to have diminished stability as the conjugation increases, in contrast to the behavior of alkenes.253 Cyclopropyl alkynes have been used as mechanistic probes to distinguish between vinyl radicals and ionic intermediates.254 Enolate radicals are also known.255
It has been postulated that the stability of free radicals is enhanced by the presence at the radical center of both an electron-donating and an electron-withdrawing group.256 This finding is called the push–pull or captodative effect(see also, Sec. 4.K.i). The effect arises from increased resonance, as in 41.
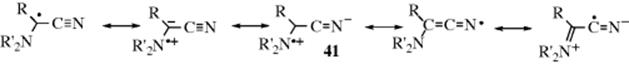
There is some evidence in favor257 of the captodative effect, some of it from ESR studies.258 However, there is also experimental259 and theoretical260 evidence against it. There is evidence that while FCH2√ and F2CH√ are more stable than CH3√, the radical CF3√ is less stable; that is, the presence of the third F destabilizes the radical.261
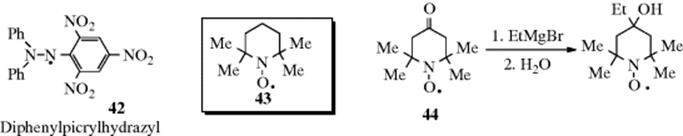
Certain radicals with unpaired electron on a carbon are also very stable.262 Radicals can be stabilized by intramolecular hydrogen bonding.263 Diphenylpicrylhydrazyl (42) is a solid that can be kept for years, and stable neutral azine radicals have been prepared.264 Nitroxide radicals were mentioned previously,265 and the commercially available TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) free radical (43) is a stable nitroxyl radical used in chemical reactions (e.g., oxidations),266 or as a spin trap.267 Nitroxyl radical 44 is a nitroxide radical so stable that reactions can be performed on it (e.g., the Grignard reaction shown with 44; see Reaction 16-24) without affecting the unpaired electron268 (the same is true for some of the chlorinated triarylmethyl radicals mentioned above269). Several nitrogen-containing groups are known to stabilize radicals, and the most effective radical stabilization is via spin delocalization.270 A number of persistent N-tert-butoxy-1-aminopyrenyl radicals (e.g., 45) have been isolated as monomeric radical crystals (see 46, the X-ray crystal
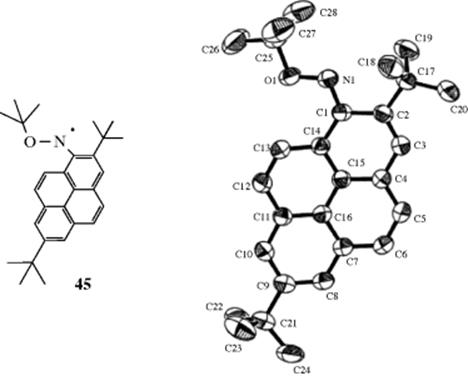
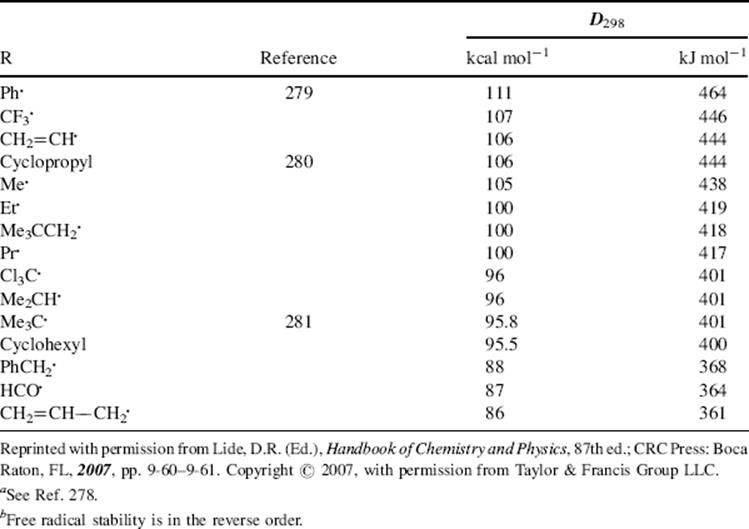
structure of 45),271 and monomeric N-alkoxyarylaminyls have been isolated.272 α-Trichloromethylbenzyl(tert-butyl)aminoxyl (47) is extremely stable.273 In aqueous media it is stable for > 30 days, and in solution in an aromatic hydrocarbon solvent it has survived for >90 days.273 Although the stable nitroxide radicals have the α-carbon blocked to prevent radical formation there, stable nitroxide radicals are also known with hydrogen at the α-carbon,274 and long-lived vinyl nitroxide radicals are known.275 A stable organic radical lacking resonance stabilization has been prepared (48), and its X-ray crystal structure was obtained.276 Dissociation energies (D values) of R–H bonds provide a measure of the relative inherent stability of free radicals R.277 Table 5.4 lists such values. 278279280281 The higher the D value, the less stable the radical. Bond-dissociation energies also have been reported for the C–H bond of alkenes and dienes282 and for the C–H bond in radical precursors XYC–H, where X,Y can be H, alkyl, COOR, COR, SR, CN, NO2, and so on.283 Bond dissociation energies for the C–O bond in hydroperoxide radicals (ROO√) have also been reported.284 However, note that basing radical stabilization energy on the difference between the bond dissociation energy (BDE) of CH3–H, as a reference point, and of R–H has been observed to have shortcomings.285 The problem is that these values are only applicable to carbon-centered radicals, and the stabilization energies are not transferable and cannot be used to estimate BDE of R–R′, R–R, or any R–X compounds.282
Table 5.4 The D298 Values for Some R–H Bondsa,b
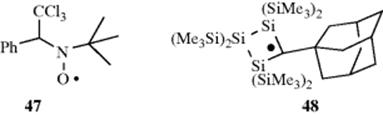
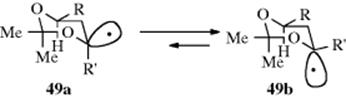
There are two possible structures for simple alkyl radicals.286 They might have sp2 bonding, in which case the structure would be planar, with the odd electron in a p orbital, or the bonding might be sp3, which would make the structure pyramidal and place the odd electron in an sp3 orbital. The ESR spectra of √CH3 and other simple alkyl radicals as well as other evidence indicate that these radicals have planar structures.287 This finding is in accord with the known loss of optical activity when a free radical is generated at a stereogenic carbon.288 In addition, electronic spectra of the CH3 and CD3 radicals (generated by flash photolysis) in the gas phase have definitely established that under these conditions the radicals are planar or near planar.289 The IR spectra of √CH3 trapped in solid argon led to a similar conclusion.290 Despite the usual loss of optical activity noted above, asymmetric radicals can be prepared in some cases. For example, asymmetric nitroxide radicals are known.291 An anomeric effect was observed in alkoxy radical 49, where the ratio of 49a/49b was 1:1.78.292
Evidence from studies on bridgehead compounds shows that although a planar configuration is more stable, pyramidal structures are not impossible. In contrast to the situation with carbocations, free radicals have often been generated at bridgeheads, although studies have shown that bridgehead free radicals are less rapidly formed than the corresponding open-chain radicals.293 In sum, the available evidence indicates that although simple alkyl free radicals prefer a planar, or near-planar shape, the energy difference between a planar and a pyramidal free radical is not great. However, free radicals in which the carbon is connected to atoms of high electronegativity (e.g., √CF3), prefer a pyramidal shape;294 increasing the electronegativity increases the deviation from planarity.295 Cyclopropyl radicals are also pyramidal.296 Free radicals with resonance are definitely planar, although triphenylmethyl-type radicals are propeller shaped,297 like the analogous carbocations (Sec. 5.A.i). Radicals possessing simple alkyl substituents attached to the radical carbon (C√) that have Csp3–Csp3 bonds, and rotation about those bonds is possible. The internal rotation barrier for the tert-butyl radical (Me3C√), for example, was estimated to be ~1.4 kcal mol−1 6 kJ mol−1.298
A number of diradicals (also called biradicals) are known,299 and the thermodynamic stability of diradicals has been examined.300 Orbital phase theory has been applied to the development of a theoretical model of localized 1,3-diradicals, and used to predict the substitution effects on the spin preference and S–T gaps, and to design stable localized carbon-centered 1,3-diradicals.301 When the unpaired electrons of a diradical are widely separated, for example, as in √CH2CH2CH2CH2√, the species behaves spectrally like two doublets. When they are close enough for interaction or can interact through an unsaturated system (as in trimethylenemethane),302 they can have total spin numbers of +1, 0, or -1, since each electron could be either ![]() or
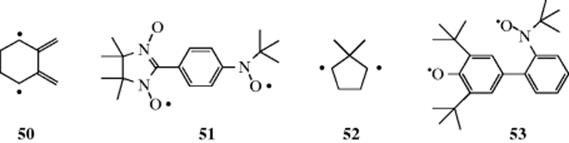
or ![]() . Spectroscopically they are called triplets,303 since each of the three possibilities is represented among the molecules and gives rise to its own spectral peak. In triplet molecules, the two unpaired electrons have the same spin. Not all diradicals have a triplet ground state. In 2,3-dimethylelecycohexane-1,4-diyl (50), the singlet and triplet states were found to be almost degenerate.304Diradicals (e.g., 51) are very stable with a triplet ground state.305 Diradicals are generally short-lived species. The lifetime of 52 was measured to be <0.1 ns and other diradicals were found to have lifetimes in the 4–316-ns range.306Diradical 53 [3,5-di-tert-butyl-3′-(N-tert-butyl-N-aminoxy)-4-oxybiphenyl] was found to have a lifetime of weeks even in the presence of oxygen, and survived brief heating in toluene up to ~60 °C.307 Radicals with both unpaired electrons on the same carbon are discussed under carbenes. 1,4-Biradicals are known, and α-carbonyl substituents increase the lifetime of the radical, and negative α-hyperconjugation (see Sec. 2.M) has been suggested as the cause.308
. Spectroscopically they are called triplets,303 since each of the three possibilities is represented among the molecules and gives rise to its own spectral peak. In triplet molecules, the two unpaired electrons have the same spin. Not all diradicals have a triplet ground state. In 2,3-dimethylelecycohexane-1,4-diyl (50), the singlet and triplet states were found to be almost degenerate.304Diradicals (e.g., 51) are very stable with a triplet ground state.305 Diradicals are generally short-lived species. The lifetime of 52 was measured to be <0.1 ns and other diradicals were found to have lifetimes in the 4–316-ns range.306Diradical 53 [3,5-di-tert-butyl-3′-(N-tert-butyl-N-aminoxy)-4-oxybiphenyl] was found to have a lifetime of weeks even in the presence of oxygen, and survived brief heating in toluene up to ~60 °C.307 Radicals with both unpaired electrons on the same carbon are discussed under carbenes. 1,4-Biradicals are known, and α-carbonyl substituents increase the lifetime of the radical, and negative α-hyperconjugation (see Sec. 2.M) has been suggested as the cause.308
5.C.ii. The Generation and Fate of Free Radicals309
Free radicals are formed from molecules by breaking a bond so that each fragment keeps one electron.310,311 The energy necessary to break the bond is supplied in one of two ways.
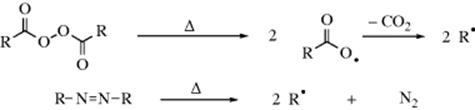
1. Thermal Cleavage. Subjection of any organic molecule to a high enough temperature in the gas phase results in the formation of free radicals. When the molecule contains bonds with D values or 20–40 kcal mol−1 (80–170 kJ mol−1), cleavage can be caused in the liquid phase. Two common examples are cleavage of diacyl peroxides to acyl radicals that decompose to alkyl radicals312 and cleavage of azo compounds to alkyl radicals313
2. Photochemical Cleavage (see Sec. 7.A.v). Light energy of 600–300 nm is 48–96 kcal mol−1 (200–400 kJ mol−1), which is on the order of magnitude of covalent-bond energies. Typical examples are photochemical cleavage of alkyl halides in the presence of triethylamine,314 alcohols in the presence of mercuric oxide and iodine,315 alkyl 4-nitrobenzenesulfenates,316 chlorine and of ketones:
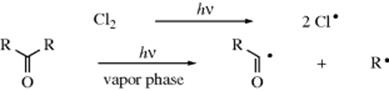
Photolytic decomposition of N-hydroxypyridin-2-thione is a method that generates hydroxyl radicals.317 The photochemistry of radicals and biradicals has been reviewed.318
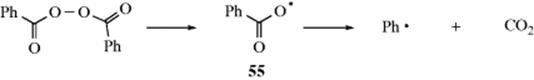
Radicals are also formed from other radicals, either by the reaction between a radical and a molecule (which must give another radical, since the total number of electrons is odd) or by cleavage of a radical319 to give another radical, for example, the decomposition of benzoyl peroxide to give the benzoyl radical:
Radicals can also be formed by oxidation or reduction, including electrolytic methods.
Reactions of free radicals either give nonradical products (termination reactions) or lead to other radicals, which themselves must usually react further (propagation reactions). The most common termination reactions are simple coupling of similar or different radicals:
![]()
Another termination process is disproportionation:320
A propagation reaction is one in which a radical reacts to give at least one radical product, which continues the radical reaction sequence. There are four principal propagation reactions, of which the first two are most common:
![]()
1. Abstraction of Another Atom or Group, Usually a Hydrogen Atom (also see Chap 14).
![]()
A radical may abstract hydrogen atoms from a second molecule, or by an intramolecular process. A bromine radical (Br√) reacts with an alkane, for example, to give HBr and a carbon radical. This type of reaction is known as hydrogen-atom transfer. Water is an excellent hydrogen atom source for many reactions involving metals.321 The reduction of a carbon radical with Bu3SnH is an example of a hydrogen-transfer reaction (see Sec. 14.A.i). Indeed, carbon radicals react as hydrogen-bond acceptors.322 Other atoms may be removed by a radical via atom-transfer reactions. A halogen atom can be transferred in some cases, including the transfer of an iodine atom from an aryl iodide to give an aryl radical.323 Solvent effects play a role in hydrogen-atom transfer (hydrogen abstraction), and hydrogen-bonding plays a role.324
2. Addition to a Multiple Bond (see Chap 15).
![]()
The radical formed from an alkene may add to the double bond of a second equivalent of alkene, and so on. This is one of the chief mechanisms for vinyl polymerization.
3. Decomposition. This process can be illustrated by the decomposition of the benzoxy radical (see 55).
4. Rearrangement.

This reaction is less common than rearrangement of carbocations, but it does occur (though not when R = alkyl or hydrogen; see Chapter 18). Perhaps the best-known rearrangement is that of cyclopropylcarbinyl radicals to a butenyl radical.325 The rate constant for this rapid ring opening has been measured in certain functionalized cyclopropylcarbinyl radicals by picosecond radical kinetics.326 Substituent effects on the kinetics of ring opening in substituted cyclopropylcarbinyl radicals have been studied.327 “The cyclopropylcarbinyl radical (56) has found an important application as a radical clock.328 Various radical processes can be clocked by the competition of direct reaction with the cyclopropylcarbinyl radical (kt) and opening of that radical to the 1-buten-4-yl radical (kr) followed by trapping. Relative rates (kt/kr) can be determined from yields of 4-X-1-butene and cyclopropylcarbinyl products as a function of the radical trap329 (X–Y) concentration. Absolute rate constants have been determined for a number of radicals with various radical traps by laser flash photolysis methods.330 From these absolute rate constants, reasonably accurate values of kt can be estimated, and with the relative rate (kt/kr), a value for kr can be calculated. From the calibrated radical-clock reaction rate (kr), rates (kt) of other competing reactions can be determined from relative rate data (kt/kr).”326 Other radical clocks are known.331
![]()
Free radicals can also be oxidized to carbocations or reduced to carbanions.332
5.C.iii. Radical Ions333
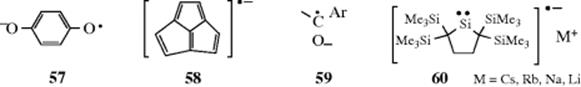
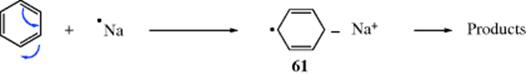
Several types of radical anions are known with the unpaired electron or the charge or both on atoms other than carbon. Examples include semiquinones334 (57), acepentalenes (58),335 ketyls336 (59) and the radical anion of the isolable dialkylsilylene (60).337 Radical anions are formed by the reaction of carbene anions with chloromethanes.338 Reactions in which alkali metals are reducing agents often involve radical anion intermediates (Birch reduction, e.g., Reaction 15-13) that proceed via radical anion 61.
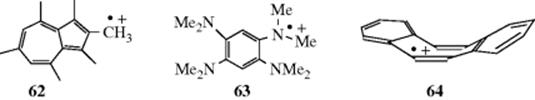
Several types of radical cation are also known.339 Typical examples include alkyl azulene cation radicals (62),340 trialkyl amine radical cations,341 1,2-bis(dialkylamino)benzenes radical cations (e.g., 63),342 dimethylsulfonium cation radicals (Me2S√+),343N-alkyl substituted imine cation radicals (Ph2C=NEt√+),344 dibenzo[a,e]cyclooctene (64, a nonplanar cation radical),345 and [n.n]paracyclophane cation radicals.346 A twisted radical cation derived from bicyclo[2.2.2]oct-2-ene has been reported.347