March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part I. Introduction
Chapter 9. Effects of Structure and Medium on Reactivity
9.D. Effect of Medium on Reactivity and Rate
There is no question that the solvent chosen for a given reaction has a profound influence on the course of that reaction. Protic versus aprotic solvents, as well as polar versus nonpolar solvents, can have effects ranging from solubility to solvent assisted ionization or stabilization of transition states. Reactions can also be done neat in one of the reactants, in the gas phase, on solid support, or in the solid phase. Environmental friendly chemistry (green chemistry) is becoming increasingly important, and chemical reactions in nonpolluting (often non-organic) solvents are of particular interest.84 This section will describe alternative reaction media as well as other medium-related things that influence chemical reactions.
9.D.i High Pressure
Acceleration of some chemical reactions is possible when high-pressure techniques are employed.85,86 The effects on a given reaction can be predicted to a certain extent because the thermodynamic properties of solutions are well known. The rate of a reaction can be expressed in terms of the activation volume, (ΔV‡)
![]()
so rate constants vary with pressure.86 “The activation volume87 is the difference in partial molal volume between the transition state and the initial state. From a synthetic point of view, this could be approximated by the molar volume.”86 If the volume of activation is negative, the rate of the reaction will be accelerated by increasing pressure. As the pressure increases, the value of ΔV‡ decreases and the system does not strictly obey the equation shown above at pressures > 10 kbar (1 bar = 0.986924 atm = 1.1019716 kg cm−2). If the transition state of a reaction involves bond formation, concentration of charge, or ionization, a negative volume of activation often results. There is a correlation between pressure and steric interactions in organic reactions.88 Cleavage of a bond, dispersal of charge, or neutralization of the transition state and diffusion control lead to a positive volume of activation. Matsumoto et al. summarized the reactions for which rate enhancement is expected at high pressure.86
1. Reactions in which the molecularity number (number of molecules) decreases when starting materials are converted to products: cycloadditions and condensations.
2. Reactions that proceed via cyclic transition states.
3. Reactions that take place through dipolar transition states.
4. Reactions with steric hindrance.
Many high-pressure reactions are done neat, but if a solvent is used, the influence of pressure on that solvent is important. The melting point generally increases at elevated pressures, and this influences the viscosity of the medium (the viscosity of liquids increases approximately two times per kilobar increase in pressure). Controlling the rate of diffusion of reactants in the medium is also important, leading to another influence of high pressure on reactivity.86,89 In most reactions, pressure is applied (5–20 kbar) at room temperature and then the temperature is increased until reaction takes place. The temperature is lowered and the pressure is reduced to isolate the products.
9.D.ii Water and Other Non-Organic Solvents
Although some reactions may be done in water,90 chemical reactions of organic substrates usually employ an organic solvent (e.g., a hydrocarbon, ether, dichloromethane, small molecular weight alcohols, and so on), but other more exotic solvents are available. For example, poly(ethylene glycol), or PEG, has been used as a solvent medium for catalytic hydrogenation (Reaction 15-11).91 For some reactions in organic solvents, the presence of water may cause unwanted side reactions, and methods have been developed to detect the presence of water in those solvents.92
With the exception of small molecular weight molecules with polar functional groups and polyfunctional molecules or salts, organic chemicals have poor solubility in water. Nonetheless, some reactions show a faster rate of reaction in water or in aqueous media.93 The first indication that water accelerated a reaction was in a patent by Hopff and Rautenstrauch in 1939,94 who reported that yields in the Diels–Alder Reaction (15-60) were enhanced in aqueous detergent solutions. In an early study, Berson et al.95 showed a clear relationship between the endo/exo product ratio and solvent polarity, in the Diels–Alder reaction of cyclopentadiene and acrylates. Breslow and Rideout.96showed there was a hydrophobic acceleration for an intermolecular Diels–Alder reaction in which cyclopentadiene reacted with methyl vinyl ketone. Clearly, there is an accelerating effect on some chemical reactions when done in water that is useful in organic chemistry.97
When nonpolar compounds are suspended in water, their relative insolubility causes them to associate, diminishing the water–hydrocarbon interfacial area (a hydrophobic effect).98 This association is greater in water than in methanol and brings the reactive partners into close proximity, increasing the rate of reaction. Any additive that increases the hydrophobic effect will increase the rate.96
Organic chemical reactions have been done is supercritical fluids, including supercritical water.99 A supercritical fluid can be either liquid or gas, but it is used in a state above the temperature and pressure where gases and liquids can coexist. The properties of a supercritical fluid are different from those of either gases or liquids under standard conditions, with no distinct liquid and gas phases at temperatures and pressures above its critical point. The critical point is the temperature, pressure, and so on, at which there are no phase boundaries. Carbon dioxide can be used as a reaction solvent when pressurized (supercritical carbon dioxide, scCO2). Carbon dioxide is nontoxic, inexpensive, abundant, and easily recycled. These properties have made it attractive as an extraction solvent.100 The low critical temperature of CO2 (Tc) 31.1 °C ensures that scCO2 is a safe solvent for many applications.101 There are solubility issues that suggest scCO2 is a rather polar solvent.102 For example, many systems with hydrocarbon chains are not very soluble in CO2.103 Water/carbon dioxide emulsions have also been employed.104 The use of (scCO2) has been explored in many reactions,105 including catalysis.106 Some applications of this technique include the electrochemical synthesis of conducting polymers107 and highly cross-linked polymers108 in scCO2, the synthesis of octyl palmitate,109 of carbonated fatty methyl esters,110 and of methyl carbamates.111 A carbonylation reaction was done is scCO2 in the course of a synthesis of trisubstituted cyclopentanes and cyclohexanes as key components of substance P antagonists.112 A continuous flow acid-catalyzed dehydration of alcohols was accomplished in scCO2.113 Supercritical fluids are playing an increasingly important role in synthetic organic chemistry.114
Other supercritical fluids can be used for chemical reactions, such as supercritical ammonia in the synthesis of labeled guanidines.115
9.D.iii Ionic Solvents
Environmentally friendly solvents,116 which include ionic liquids, are of great interest.117 An ionic liquid is a salt in which the ions are poorly coordinated, usually leading to their being liquid at <100 °C and sometimes at room temperature.118 In such ionic species, there is usually at least one ion with a delocalized charge, whereas the other component is usually organic. This combination inhibits the formation of a stable crystal lattice. The structure and solvation properties of solutes in ionic liquids have been studied.119 It was discovered that some ionic liquids are suitable as a medium for chemical reactions.120 Both methylimidazolium and pyridinium ions form the basis of common ionic liquids that have been used in organic chemistry.121 One of the most common ionic liquids used as a solvent is 1-butyl-3-methylimidazolium as the hexafluorophosopahte, (10, Bmim PF6).122Hydrogenbutylimidazolium tetrafluoroborate (HBuIm, 11) and 1,3-dibutylimidazolium, tetrafluoroborate (DiBuIm, 12), for example,123 have been reported to facilitate Diels–Alder Reactions (15-60).124 It is known that a proton on C-2 of imidazolium cations (e.g., 10-12) is relatively acidic.125 Carbene formation is common and the anion generated by treatment with base can undergo substitution reactions.125 These facts lead to a caution that undesired side reactions are possible when these ionic liquids are employed as solvents.125,126 Pyridinium-based ionic liquids [e.g., ethylpyridinium tetrafluoroborate (13)] have also been used.127 Several room temperature ionic liquids have been synthesized from amino acids.128
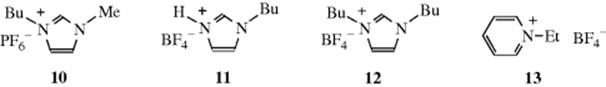
Ionic solvents have been used to facilitate heterocyclic reactions,129 several catalytic reactions,130 the Heck Reaction (13-9)131 and other Pd catalyzed C–C bond-forming reactions,132 the oxidation of alcohols with hypervalent iodine reagents (19-3),133 and the catalytic asymmetric dihydroxylation of alkenes (15-48) using a recoverable and reusable Os/ligand complex.134 The camphorsulfonate anion has been used as a counterion for imidazolium salts, and shown to increase the number of unsolvated imidazolium cations.135 This ionic liquid was then shown to influence the endo/exo ratio in a stereoselective Diels–Alder Reaction (15-60).135 Other catalytic reactions in ionic liquids are known.136 Other chiral ionic liquids are known.137 Reactions performed in an ionic liquid are a rapidly growing area of organic chemistry, and has been expanded to include microwave reactions (see Sec. 7.C) in ionic solvents.138The development and use of ionic solvents is a growth area of organic chemistry.139 Also note that some ionic liquids are categorized as Lewis base (Sec. 8.E), which will influence the acidity of dissolved compounds.140 There are also acidic Br![]() nsted ionic liquids.141
nsted ionic liquids.141
9.D.iv. Solventless Reactions
In some cases, it should be possible to accomplish a chemical transformation without the use of a solvent. Dry media reaction under microwave irradiation is an important area of study (see Sec. 7.C).142 There are several advantages of solventless reactions: (1) the possibility of direct formation of high-purity compounds, (2) the possibility of sequential reactions, (3) fast kinetics, (4) lower energy usage, (5) minimal need for preformed salts and metal–metalloid complexes, (6) simplicity and low equipment cost, and (7) the possibility of avoiding functional group protection–deprotection.143 Potential difficulties include the possibility of hot spots and runaway reactions, and difficulties in handling solid or highly viscous materials.144 An example of this approach is the aldol condensation, where a single aldol product was obtained in high yield.145 3-Carboxylcoumarins have been produced via a solventless aldol.143
Notes
1. See Klumpp, G.W. Reactivity in Organic Chemistry, Wiley, NY, 1982. For a general theoretical approach to organic reactivity, see Pross, A. Adv. Phys. Org. Chem. 1985, 21, 99.
2. See Topsom, R.D. Prog. Phys. Org. Chem. 1987, 16, 125, Mol. Struct. Energ. 1987, 4, 235.
3. Hughes, E.D. Q. Rev. Chem. Soc. 1948, 2, 107.
4. Brown, H.C. Boranes in Organic Chemistry, Cornell University Press, Ithaca, NY, 1972, pp. 114–121.
5. See Stirling, C.J.M. Tetrahedron 1985, 41, 1613; Pure Appl. Chem. 1984, 56, 1781.
6. Brown, H.C.; Fletcher, R.S. J. Am. Chem. Soc. 1949, 71, 1845.
7. Bartlett, P.D.; Tidwell, T.T. J. Am. Chem. Soc. 1968, 90, 4421.
8. See Brown, H.C. Boranes in Organic Chemistry, Cornell University Press, Ithaca, NY, 1972, pp. 105–107, 126–128.
9. Brown, H.C.; Borkowski, M. J. Am. Chem. Soc. 1952, 74, 1894. See also, Brown, H.C.; Ravindranathan, M.; Peters, E.N.; Rao, C.G.; Rho, M.M. J. Am. Chem. Soc. 1977, 99, 5373.
10. See Schneider, H.; Thomas, F. J. Am. Chem. Soc. 1980, 102, 1424.
11. Sands, R.D. J. Org. Chem. 1994, 59, 468.
12. See Green, B.S.; Arad-Yellin, R.; Cohen, M.D. Top. Stereochem. 1986, 16, 131; Öki, M. Acc. Chem. Res. 1984, 17, 154; Seeman, J.I. Chem. Rev. 1983, 83, 83. See also, Öki, M.; Tsukahara, J.; Moriyama, K.; Nakamura, N. Bull. Chem. Soc. Jpn. 1987, 60, 223, and other papers in this series.
13. Fodor, G.; Bruckner, V.; Kiss, J.; Óhegyi, G. J. Org. Chem. 1949, 14, 337.
14. See Eliel, E.L. Stereochemistry of Carbon Compounds, McGraw-Hill, NY, 1962, pp. 219–234.
15. Barton, D.H.R.; McCapra, F.; May, P.J.; Thudium, F. J. Chem. Soc. 1960, 1297.
16. See Exner, O. Correlation Analysis of Chemical Data, Plenum, NY, 1988; Johnson, C.D. The Hammett Equation, Cambridge University Press, Cambridge, 1973; Shorter, J. Correlation Analysis of Organic Reactivity, Wiley, NY, 1982; Chapman, N.B.; Shorter, J. Correlation Analysis in Chemistry: Recent Advances, Plenum, NY, 1978. Also see Connors, K.A. Chemical Kinetics, VCH, NY, 1990, pp. 311–383; Lewis, E.S. in Bernasconi, C.F. Investigation of Rates and Mechanisms of Reactions (Vol. 6 of Weissberger, A. Techniques of Chemistry), 4th ed., Wiley, NY, 1986, pp. 871–901; Jones, R.A.Y. Physical and Mechanistic Organic Chemistry, 2nd ed., Cambridge University Press, Cambridge, 1984, pp. 38–68; Hine, J. Structural Effects in Organic Chemistry, Wiley, NY, 1975, pp. 55–102. For a historical perspective, see Grunwald, E. CHEMTECH 1984, 698.
17. For a review, see Jaffé, H.H. Chem. Rev. 1953, 53, 191.
18. Additional ρ values are given in Wells, P.R. Chem. Rev. 1963, 63, 171 and van Bekkum, H.; Verkade, P.E.; Wepster, B.M. Recl. Trav. Chim. Pays-Bas 1959, 78, 821.
19. For a review of Hammett treatment of NMR chemical shifts, see Ewing, D.F. in Chapman, N.B.; Shorter, J. Correlation Analysis in Chemistry: Recent Advances, Plenum, NY, 1978, pp. 357–396.
20. Unless otherwise noted, σ values are from Exner, O. in Chapman, N.B.; Shorter, J. Correlation Analysis in Chemistry: Recent Advances, Plenum, NY, 1978, pp. 439–540, and σ+ values from Okamoto, Y.; Inukai, T.; Brown, H.C. J. Am. Chem. Soc. 1958, 80, 4969; Brown, H.C.; Okamoto, Y. J. Am. Chem. Soc. 1958, 80, 4979. σ− values, except as noted, are from Jaffé, H.H. Chem. Rev. 1953, 53, 191. Also see Hansch, C.; Leo, A.; Taft, R.W. Chem. Rev. 1991, 91, 165; Egorochkin, A.N.; Razuvaev, G.A. Russ. Chem. Rev. 1987, 56, 846. For values for heteroaromatic groups, see Mamaev, V.P.; Shkurko, O.P.; Baram, S.G. Adv. Heterocycl. Chem. 1987, 42, 1.
24. Hine, J. J. Am. Chem. Soc. 1960, 82, 4877; Jones, R.A.Y. Physical and Mechanistic Organic Chemistry, 2nd ed., Cambridge Univ. Press, Cambridge, 1984, p. 42.
25. See Hine, J. J. Am. Chem. Soc. 1960, 82, 4877.
26. Matsui, T.; Ko, H.C.; Hepler, L.G. Can. J. Chem. 1974, 52, 2906.
27. de la Mare, P.B.D.; Newman, P.A. Tetrahedron Lett. 1982, 23, 1305 give this value as –1.6.
28. Amin, H.B.; Taylor, R. Tetrahedron Lett. 1978, 267.
29. Sjöström, M.; Wold, S. Chem. Scr. 1976, 9, 200.
30. Byrne, C.J.; Happer, D.A.R.; Hartshorn, M.P.; Powell, H.K.J. J. Chem. Soc. Perkin Trans. 2 1987, 1649.
31. For a review of directing and activating effects of C=O, C=C, C=N, and C=S groups, see Charton, M. in Patai, S. The Chemistry of Double-Bonded Functional Groups, Vol. 2, pt. 1, Wiley, NY, 1989, pp. 239–298.
32. For a review of directing and activating effects of C![]() N and C
N and C![]() C groups, see Charton, M. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement C, pt. 1, Wiley, NY, 1983, pp. 269–323.
C groups, see Charton, M. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement C, pt. 1, Wiley, NY, 1983, pp. 269–323.
33. McDaniel, D.H.; Brown, H.C. J. Org. Chem. 1958, 23, 420.
34. Ustynyuk, Yu.A.; Subbotin, O.A.; Buchneva, L.M.; Gruzdneva, V.N.; Kazitsyna, L.A. Doklad. Chem. 1976, 227, 175.
35. Lewis, E.S.; Johnson, M.D. J. Am. Chem. Soc. 1959, 81, 2070.
21. See Dubois, J.E.; Ruasse, M.; Argile, A. J. Am. Chem. Soc. 1984, 106, 4840; Ruasse, M.; Argile, A.; Dubois, J.E. J. Am. Chem. Soc. 1984, 106, 4846; Lee, I.; Shim, C.S.; Chung, S.Y.; Kim, H.Y.; Lee, H.W. J. Chem. Soc. Perkin Trans. 2 1988, 1919.
22. Hine, J. J. Am. Chem. Soc. 1960, 82, 4877.
23. Binev, I.G.; Kuzmanova, R.B.; Kaneti, J.; Juchnovski, I.N. J. Chem. Soc. Perkin Trans. 2 1982, 1533.
36. Stone, R.M.; Pearson, D.E. J. Org. Chem. 1961, 26, 257.
37. Berliner, E.; Winikov, E.H. J. Am. Chem. Soc. 1959, 81, 1630; See also, Well, P.R.; Ehrenson, S.; Taft, R.W. Prog. Phys.Org. Chem. 1968, 6, 147.
38. See Charton, M. in Chapman, N.B.; Shorter, J. Correlation Analysis in Chemistry: Recent Advances, Plenum, NY, 1978, pp. 175–268; Tomasik, P.; Johnson, C.D. Adv. Heterocycl. Chem. 1976, 20, 1.
39. See Ford, G.P.; Katritzky, A.R.; Topsom, R.D. in Correlation Analysis in Chemistry: Recent Advances, Plenum, NY, ,1978, pp. 269–311; Charton, M. Prog. Phys. Org. Chem. 1973, 10, 81.
40. See Exner, O. Prog. Phys. Org. Chem. 1990, 18, 129.
41. For reviews of the separation of resonance and field effects, see Charton, M. Prog. Phys. Org. Chem. 1981, 13, 119; Shorter, J. Q. Rev. Chem. Soc. 1970, 24, 433; Chem. Ber. 1969, 5, 269. For a review of field and inductive effects, see Reynolds, W.F. Prog. Phys. Org. Chem. 1983, 14, 165. For a review of field effects on reactivity, see Grob, C.A. Angew. Chem. Int. Ed. 1976, 15, 569.
42. Ingold, C.K. J. Chem. Soc. 1930, 1032.
43. Also see Draffehn, J.; Ponsold, K. J. Prakt. Chem. 1978, 320, 249.
44. The symbol σF is also used in the literature; sometimes in place of σI, and sometimes to indicate only the field (not the inductive) portion of the total effect (Sec. 1.G).
45. There is another set of values (called σ∗ values) that are also used to correlate field effects. These are related to σI values by σI(X) = 0.45σ. Only σI, and not σ∗ values are discussed.
46. Wells, P.R. Chem. Rev. 1963, 63, 171, p. 196.
47. These values are from Bromilow, J.; Brownlee, R.T.C.; Lopez, V.O.; Taft, R.W. J. Org. Chem. 1979, 44, 4766, but the values for NHAc, OH, and I are from Wells, P.R.; Ehrenson, S.; Taft, R.W. Prog. Phys. Org. Chem. 1968, 6, 147, the values for Ph and NMe3+ are from Taft, R.W.; Ehrenson, S.; Lewis, I.C.; Glick, R. J. Am.Chem. Soc. 1959, 81, 5352; Taft, R.W.; Deno, N.C.; Skell, P.S. Annu. Rev. Phys. Chem. 1958, 8, 287, and the value for CMe3 is from Seth-Paul, W.A.; de Meyer-van Duyse, A.; Tollenaere, J.P. J. Mol. Struct. 1973, 19, 811. The values for the CH2Ph and CH2COCH3 groups were calculated from σ∗ values by the formula given in ref. 45. Also see Charton, M. Prog. Phys. Org. Chem. 1981, 13, 119; Taylor, P.J.; Wait, A.R. J. Chem. Soc. Perkin Trans. 2 1986, 1765.
48. For ![]() values for some other NR2 groups, see Korzhenevskaya, N.G.; Titov, E.V.; Chotii, K.Yu.; Chekhuta, V.G. J. Org. Chem. USSR 1987, 28, 1109.
values for some other NR2 groups, see Korzhenevskaya, N.G.; Titov, E.V.; Chotii, K.Yu.; Chekhuta, V.G. J. Org. Chem. USSR 1987, 28, 1109.
49. It has been shown that charged groups (called polar substituents) cannot be included with uncharged groups (dipolar substituents) in one general scale of electrical substituent effects: Marriott, S.; Reynolds, J.D.; Topsom, R.D. J. Org. Chem. 1985, 50, 741.
50. Taft, R.W. J. Phys. Chem. 1960, 64, 1805; Taft, R.W.; Lewis, I.C. J. Am. Chem. Soc. 1958, 80, 2436; Taft, R.W.; Deno, N.C.; Skell, P.S. Annu. Rev. Phys. Chem. 1958, 9, 287, see pp. 290–293.
51. Ehrenson, S.; Brownlee, R.T.C.; Taft, R.W. Prog. Phys. Org. Chem. 1973, 10, 1. See also, Taft, R.W.; Topsom, R.D. Prog. Phys. Org. Chem. 1987, 16, 1; Charton, M. Prog. Phys. Org. Chem. 1987, 16, 287.
52. Taft, R.W.; Lewis, I.C. J. Am. Chem. Soc. 1959, 81, 5343; Reynolds, W.F.; Dais, P.; MacIntyre, D.W.; Topsom, R.D.; Marriott, S.; von Nagy-Felsobuki, E.; Taft, R.W. J. Am. Chem. Soc. 1983, 105, 378.
53. Also see Happer, D.A.R.; Wright, G.J. J. Chem. Soc. Perkin Trans. 2 1979, 694.
54. Taft, R.W.; Ehrenson, S.; Lewis, I.C.; Glick, R.E. J. Am. Chem. Soc. 1959, 81, 5352.
55. Bromilow, J.; Brownlee, R.T.C.; Lopez, V.O.; Taft, R.W. J. Org. Chem. 1979, 44, 4766. See also, Marriott, S.; Topsom, R.D. J. Chem. Soc. Perkin Trans. 2 1985, 1045.
56. For a set of σR values for use in XY+ systems, see Charton, M. Mol. Struct. Energ. 1987, 4, 271.
57. See de Ligny, C.L.; van Houwelingen, H.C. J. Chem. Soc. Perkin Trans. 2 1987, 559.
58. See Shorter, J. in Chapman, N.B.; Shorter, J. Advances in Linear Free Energy Relationships, Plenum, NY, 1972, pp. 98–103.
59. See Screttas, C.G. J. Org. Chem. 1979, 44, 3332; Hanson, P. J. Chem. Soc. Perkin Trans. 2 1984, 101.
60. See DeTar, D.F. J. Org. Chem. 1980, 45, 5166; J. Am. Chem. Soc. 1980, 102, 7988.
61. See Shorter, J. in Chapman, N.B.; Shorter, J. Correlation Analysis in Chemistry: Recent Advances, Plenum, NY, 1978, pp. 119–173, pp. 126–144; Afanas'ev, I.B. J. Chem. Soc. Perkin Trans. 2 1984, 1589; Ponec, R. Coll. Czech. Chem. Commun. 1983, 48, 1564.
62. Swain, C.G.; Unger, S.H.; Rosenquist, N.R.; Swain, M.S. J. Am. Chem. Soc. 1983, 105, 492 and references cited therin.
63. From Swain, C.G.; Unger, S.H.; Rosenquist, N.R.; Swain, M.S. J. Am. Chem. Soc. 1983, 105, 492. Also see Hansch, C.; Leo, A.; Taft, R.W. Chem. Rev. 1991, 91, 165.
64. The Swain-Lupton treatment has been criticized by Reynolds, W.F.; Topsom, R.D. J. Org. Chem. 1984, 49, 1989; Hoefnagel, A.J.; Oosterbeek, W.; Wepster, B.M. J. Org. Chem. 1984, 49, 1993; Charton, M. J. Org. Chem.1984, 49, 1997. For a reply, see Swain, C.G. J. Org. Chem. 1984, 49, 2005. See Charton, M. Prog. Phys. Org. Chem. 1981, 13, 119; Nakazumi, H.; Kitao, T.; Zollinger, H. J. Org. Chem. 1987, 52, 2825.
65. See Gallo, R.; Roussel, C.; Berg, U. Adv. Heterocycl. Chem. 1988, 43, 173; Gallo, R. Prog. Phys. Org. Chem. 1983, 14, 115; Unger, S.H.; Hansch, C. Prog. Phys. Org. Chem. 1976, 12, 91.
66. Also see De Tar, D.F.; Delahunty, C. J. Am. Chem. Soc. 1983, 105, 2734.
67. See McClelland, R.A.; Steenken, S. J. Am. Chem. Soc. 1988, 110, 5860.
68. Taken from Gallo, R.; Roussel, C.; Berg, U. Adv. Heterocycl. Chem. 1988, 43, 173; Gallo, R. Prog. Phys. Org. Chem. 1983, 14, 115; Unger, S.H.; Hansch, C. Prog. Phys. Org. Chem. 1976, 12, 91. Charton, M. J. Org. Chem.1976, 41, 2217; and Meyer, A.Y. J. Chem. Soc. Perkin Trans. 2 1986, 1567.
69. In Taft's original work, Me was given the value 0. The Es values in Table 9.7 can be converted to the orginal values by adding 1.24.
70. Charton, M. J. Am. Chem. Soc. 1969, 91, 615.
71. Charton, M. J. Am. Chem. Soc. 1975, 97, 1552; J. Org. Chem. 1976, 41, 2217. See also, Charton, M. J. Org. Chem. 1978, 43, 3995; Idoux, J.P.; Schreck, J.O. J. Org. Chem. 1978, 43, 4002.
72. Meyer, A.Y. J. Chem. Soc. Perkin Trans. 2 1986, 1567.
73. See DeTar, D.F. J. Org. Chem. 1980, 45, 5166; J. Am. Chem. Soc. 1980, 102, 7988.
74. MacPhee, J.A.; Panaye, A.; Dubois, J.E. J. Org. Chem. 1980, 45, 1164; Dubois, J.E.; MacPhee, J.A.; Panaye, A. Tetrahedron 1980, 36, 919. See also, Datta, D.; Sharma, G.T. J. Chem. Res. (S) 1987, 422.
75. Fellous, R.; Luft, R. J. Am. Chem. Soc. 1973, 95, 5593.
76. Komatsuzaki, T.; Sakakibara, K.; Hirota, M. Tetrahedron Lett. 1989, 30, 3309; Chem. Lett. 1990, 1913.
77. Beckhaus, H. Angew. Chem. Int. Ed. 1978, 17, 593.
78. See Fujita, T.; Nishioka, T. Prog. Phys. Org. Chem. 1976, 12, 49; Charton, M. Prog. Phys. Org. Chem. 1971, 8, 235. See also, Robinson, C.N.; Horton, J.L.; Fosheé, D.O.; Jones, J.W.; Hanissian, S.H.; Slater, C.D. J. Org. Chem. 1986, 51, 3535.
79. This is not the same as the ortho effect discussed in Section 11.B.iv.
80. Charton, M. Can. J. Chem. 1960, 38, 2493.
81. See Schreck, J.O. J. Chem. Educ. 1971, 48, 103.
82. See, however, Gawley, R.E. J. Org. Chem. 1981, 46, 4595.
83. Also see Williams, A. Acc. Chem. Res. 1984, 17, 425.
84. Clark, J.H. Green Chem. 1999, 1, 1; Cave, G.W.V.; Raston, C.L.; Scott, J.L. Chem. Commun. 2001, 2159.
85. Jenner, G. Tetrahedron 2002, 58, 5185; Matsumoto, K.; Morris, A.R. Organic Synthesis at High Pressure, Wiley, New York, 1991.
86. Matsumoto, K.; Sera, A.; Uchida, T. Synthesis 1985, 1; Matsumoto, K.; Sera, A. Synthesis. 1985, 999. Also see Benito-López, F.; Egberink, R.J.M.; Reinhoudt, D.N.; Verboom, W. Tetrahedron 2008, 64, 10023.
87. See le Noble, W.J. Progr. Phys. Org. Chem. 1967, 5, 207; Isaacs, N.S. Liquid Phase High Pressure Chemistry, Wiley, Chichester, 1981; Asano, T.; le Noble, W.J. Chem. Rev. 1978,78, 407.
88. Jenner, G. Tetrahedron 2005, 61, 3621.
89. Firestone, R.A.; Vitale, M.A. J. Org. Chem. 1981, 46, 2160.
90. Organic Reactions in Water: Principles, Strategies and Applications, Lindström, U.M. (Ed.), Blackwell, Oxford, 2007; Chanda, A.; Fokin, V.V. Chem. Rev. 2009, 109, 725.
91. Chandrasekhar, S.; Prakash, S.J.; Rao, C.L. J. Org. Chem. 2006, 71, 2196. PEG has also been used for the synthesis of β-amino sulfides. See Kamal, A.; Reddy, D.R.; Rajendar Tetrahedron Lett. 2006, 47, 2261.
92. Sun, H.; Wang, B.; DiMagno, S.G. Org. Lett. 2008, 10, 4413.
93. See Pirrung, M.C. Chemistry: European J. 2006, 12, 1312.
94. Hopff, H.; Rautenstrauch, C.W. U.S. Patent 2,262,002, 1939 [Chem. Abstr. 36: 10469, 1942].
95. Berson, J.A.; Hamlet, Z.; Mueller, W.A. J. Am. Chem. Soc. 1962, 84, 297.
96. Rideout, D.; Breslow, R. J. Am. Chem. Soc. 1980, 102, 7816.
97. Engberts, J.B.F.N.; Blandamer, M.J. Chem. Commun. 2001, 1701; Lindström, U.M. Chem. Rev. 2002, 102, 2751; Ribe, S.; Wipf, P. Chem. Commun. 2001, 299.
98. For a review of chemical reactions in aqueous media with a focus on C–C bond formation, see Li, C.-J. Chem. Rev. 2005, 105, 3095. For microwave assisted synthesis in water, see Dallinger, D.; Kappe, C.O. Chem. Rev. 2007, 107, 2563.
99. Weingärtner, H.; Franck, E.U. Angew. Chem. Int. Ed.2005, 44, 2672; Fraga-Dubreuil, J.; Poliakoff, M. Pure Appl. Chem. 2006, 78,1971.
100. See Raynie, D.E. Anal. Chem. 2004, 76, 4659.
101. Subramaniam, B.; Rajewski, R. A.; Snavely, K. J. Pharm. Sci. 1997, 86, 885.
102. Raveendran, P.; Ikushima, Y.; Wallen, S.L. Acc. Chem. Res. 2005, 38, 478.
103. Consani, K.A.; Smith, R.D.J. Supercrit. Fluids 1990, 3, 51.
104. Jacobson, G.B.; Lee, Jr., C.T.; da Rocha, S.R.P.; Johnston, K.P. J. Org. Chem. 1999, 64, 1207; Jacobson, G.B.; Lee, Jr., C.T.; Johnston, K.P. J. Org. Chem. 1999, 64, 1201.
105. Gopalan, A.D.; Wai, C.M.; Jacobs, H.K. Supercritical Carbon Dioxide: Separations and Processes, American Chemical Society (distributed by Oxford University Press), Washington, DC. 2003; Beckman, E.J. Ind. Eng. Chem. Res. 2003, 42, 1598; Wang, S.; Kienzle, F. Ind. Eng. Chem. Res. 2000, 39, 4487.
106. Leitner, W. Acc. Chem. Res. 2002, 35, 746.
107. Anderson, P.E.; Badlani, R.N.; Mayer, J.; Mabrouk, P.A. J. Am. Chem. Soc. 2002, 124, 10284.
108. Cooper, A.I.; Hems, W.P.; Holmes, A.B. Macromolecules 1999, 32, 2156.
109. Madras, G.; Kumar, R.; Modak, J. Ind. Eng. Chem. Res. 2004, 43, 7697,1568.
110. Doll, K.M.; Erhan, S.Z. J. Agric. Food Chem. 2005, 53, 9608.
111. Selva, M.; Tundo, P.; Perosa, A.; Dall'Acqua, F. J. Org. Chem. 2005, 70, 2771.
112. Kuethe, J.T.; Wong, A.; Wu, J.; Davies, I.W.; Dormer, P.G.; Welch, C.J.; Hillier, M.C.; Hughes, D.L.; Reider, P.J. J. Org. Chem. 2002, 67, 5993.
113. Gray, W.K.; Smail, F.R.; Hitzler, M.G.; Ross, S.K.; Poliakoff, M. J. Am. Chem. Soc. 1999, 121, 10711.
114. See Prajapati, D.; Gohain, M. Tetrahedron 2004, 60, 815.
115. Jacobson, G.B.; Westerberg, G.; Markides, K.E.; Langstrom, B. J. Am. Chem. Soc. 1996, 118, 6868.
116. Alternative Solvents for Green Chemistry, Kerton, F.M.; Clark J.M.; Kraus, G.A. Royal Society of Chemistry, Cambridge, 2009.
117. But also see Scammells, P.J.; Scott, J.L.; Singer, R.D. Austr. J. Chem. 2005, 58, 155.
118. For a discussion of physical properties, see Ludwig, R.; Kragl, U. Angew. Chem. Int. Ed. 2007, 46, 6582.
119. Hardacre, C.; Holbrey, J.D.; Nieuwenhuyzen, M.; Youngs, T.G.A. Acc. Chem. Res. 2007, 40, 1146; Greaves, T.L.; Drummond, C.J. Chem. Rev. 2008, 108, 206. See also Lungwitz, R.; Strehmel, V.; Spange, S. New J. Chem.2010, 34, 1135.
120. Wasserscheid, P.; Keim, W. Angew. Chem. Int. Ed. 2000, 39, 3772; Earle, M.J.; Seddon, K.R. Pure. Appl. Chem. 2000, 72, 1391; Ionic Liquids in Synthesis, Wasserscheid, P.; Welton, T.; Wiley–VCH, NY, 2002; Chemistry in Alternative Reaction Media, Adams, D.J.; Dyson, P.J.; Taverner, S.J.; Wiley, 2003. For a discussion of the solvating ability, see Chiappe, C.; Malvaldi, M.; Pomelli, C.S. Pure Appl. Chem. 2009, 81, 767.
121. Rogers, R.D.; Voth, G.A. Acc. Chem. Res. 2007, 40,1077;
122. Dupont, J.; Consorti, C.S.; Suarez, P.A.Z.; de Souza, R.F. Org. Synth. Coll. Vol. X, 184.
123. For discussion of HBuIM and DiBuIm, see Harlow, K.J.; Hill, A.F.; Welton, T. Synthesis 1996, 697; Holbrey, J.D.; Seddon, K.R. J. Chem. Soc., Dalton Trans. 1999, 2133; Larsen, A.S.; Holbrey, J.D.; Tham, F.S.; Reed, C.A. J. Am. Chem. Soc. 2000, 122, 7264.
124. Jaegar, D.A.; Tucker, C.E. Tetrahedron Lett. 1989, 30, 1785.
125. Handy, S.T.; Okello, M. J. Org. Chem. 2005, 70, 1915.
126. For a discussion of the reactivity of ionic liquids, see Chowdhury, S.; Mohan, R.S.; Scott, J.L. Tetrahedron 2007, 63, 2363.
127. See Xiao, Y.; Malhotra, S.V. Tetrahedron Lett. 2004, 45, 8339.
128. Fukumoto, K.; Yoshizawa, M.; Ohno, H. J. Am. Chem. Soc. 2005, 127, 2398. Also see Chen, X.; Li, X.; Hu, A.; Wang, F. Tetrahedron Asymmetry 2008, 19, 1.
129. Martins, M.A.P.; Frizzo, C.P.; Moreira, D.N.; Zanatta, N.; Bonacorso, H.G. Chem. Rev. 2008, 108, 2015.
130. See Toma, Š.; Me![]() iarová. M.; Šebesta, R. Eur. J. Org. Chem. 2009, 321.
iarová. M.; Šebesta, R. Eur. J. Org. Chem. 2009, 321.
131. Handy, S.T.; Okello, M.; Dickenson, G. Org. Lett. 2003, 5, 2513.
132. Calò, V.; Nacci, A.; Monopoli, A. Eur. J. Org. Chem. 2006, 3791.
133. Yadav, J.S.; Reddy, B.V.S.; Basak, A.K.; Narsaiah, A.V. Tetrahedron 2004, 60, 2131.
134. Branco, L.C.; Afonso, C.A.M. J. Org. Chem. 2004, 69, 4381.
135. Nobuoka, K.; Kitaoka, S.; Kunimitsu, K.; Iio, M.; Harran, T.; Wakisaka, A.; Ishikawa, Y. J. Org. Chem. 2005, 70, 10106.
136. Pârvulescu, V.I.; Hardacre, C. Chem. Rev. 2007, 107, 2615.
137. Baudequin, C.; Brégeon, D.; Levillain, J.; Guillen, F.; Plaquevent, J.-C.; Gaumont, A.C. Tetrahedron Asymmetry 2005, 16, 3921; Pernak, J.; Feder-Kubis, J. Tetrahedron Asymmetry 2006, 17, 1728; Luo, S.-P.; Xu, D.-Q.; Yue, H.-D.; Wang, L.-P.; Yang, W.-L.; Xu, Z.-Y. Tetrahedron Asymmetry 2006, 17, 2028.
138. See Leadbeater, N.E.; Torenius, H.M. J. Org. Chem. 2002, 67, 3145.
139. For studies to expand the polarity range of ionic solvents see Dzyuba, S.V.; Bartsch, R.A. Tetrahedron Lett. 2002, 43, 4657. See Ionic Liquids: From Knowledge to Application, Plechkova, N.V.; Rogers, R.D.; Seddon, K.R. (Eds.), American Chemical Society, Washington, DC (distributed by Oxford University Press), 2010.
140. MacFarlane, D.R.; Pringle, J.M.; Johansson, K.M.; Forsyth, S.A.; Forsyth, M. Chem. Commun. 2006, 1905.
141. Hajipour, A.R.; Rafiee, F. Org. Prep. Proceed. Int. 2010, 42, 285.
142. Kidwai, M. Pure Appl. Chem. 2001, 73, 147.
143. Cave, G.W.V.; Raston, C.L.; Scott, J.L. Chem. Commun. 2001, 2159; Toda, F.; Tanaka, K. Chem. Rev. 2000, 100, 1025.
144. Raston, C.L. Chemistry in Australia 2004, 10.
145. Toda, F.; Tanaka, K.; Hamai, K. J. Chem. Soc., Perkin Trans. 1 1990, 3207.