March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 10. Aliphatic Substitution, Nucleophilic and Organometallic
10.C. The Neighboring-Group Mechanism113
It is occasionally found with certain substrates that (1) the rate of reaction is greater than expected, and (2) the configuration at a chiral carbon is retained and not inverted or racemized. In these cases, there is usually a group with an unshared pair of electrons β to the leaving group (or sometimes farther away). The mechanism operating in such cases is called the neighboring-group mechanism and consists essentially of two SN2 substitutions, each causing an inversion so the net result is retention of configuration.114 In the first step of this reaction, the neighboring group acts as a nucleophile, pushing out the leaving group, but still retaining attachment to the molecule. In the second step, the external nucleophile displaces the neighboring group by a backside attack:
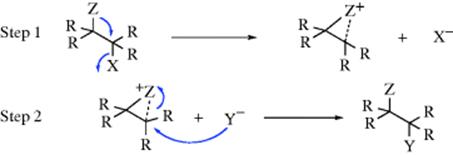
The reaction obviously must go faster than if Y were attacking directly, since if the latter process were faster, it would be happening. The neighboring group Z is said to be lending anchimeric assistance. The rate law followed in the neighboring-group mechanism is the first-order law shown in Eq. (10-2) or (10-3)); that is, Y does not take part in the rate-determining step.
The reason attack by Z is faster than that by Y is that the group Z is more available. In order for Y to react, it must collide with the substrate, but Z is immediately available by virtue of its position. A reaction between the substrate and Y involves a large decrease in entropy of activation (ΔS‡), since the reactants are far less free in the transition state than before. Reaction of Z involves a much smaller loss of ΔS‡ (see Sec. 6.D).115
It is not always easy to determine when a reaction rate has been increased by anchimeric assistance. In order to be certain, it is necessary to know what the rate would be without participation by the neighboring group. An obvious way to examine this question is to compare the rates of the reaction with and without the neighboring group, (e.g., HOCH2CH2Br vs CH3CH2Br). However, this will certainly not give an accurate determination of the extent of participation, since the steric and field effects of H and OH are not the same. Furthermore, no matter what the solvent, the shell of solvent molecules that surrounds the polar protic OH group must differ greatly from that which surrounds the nonpolar H. Because of these considerations, it is desirable to have a large increase in the rate, preferably > 50-fold, before a rate increase is attributed to neighboring-group participation.
The first important evidence for the existence of this mechanism was the demonstration that retention of configuration can occur if the substrate is suitable. It was shown that the threo dl pair of 3-bromo-2-butanol when treated with HBr gave dl-2,3-dibromobutane, while the erythro pair gave the meso isomer (22).116
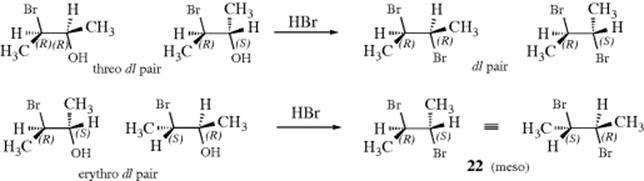
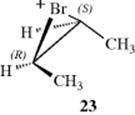
This result indicated that retention had taken place. Note that both products are optically inactive and so cannot be told apart by differences in rotation. The meso and dl dibromides have different boiling points and indexes of refraction and were identified by these properties. Even more convincing evidence was that either of the two threo isomers alone gave not just one of the enantiomeric dibromides, but the dl pair. The reason for this is that the intermediate present after the attack by the neighboring group (23) is symmetrical, so the external nucleophile Br− can attack both carbon atoms equally well. Intermediate 23 is a bromonium ion, the existence of which has been demonstrated in several types of reactions (see Reaction 15-39).
Although 23 is symmetrical, intermediates in most neighboring-group mechanisms are not, and it is therefore possible to get not a simple substitution product, but a rearrangement. This will happen if Y attacks not the carbon atom from which X left, but the one to which Z was originally attached:
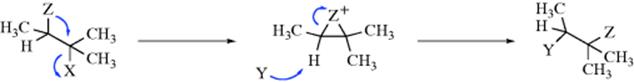
In such cases, substitution and rearrangement products are often produced together. For a discussion of rearrangements, see Chapter 18.
Another possibility is that the intermediate may be stable or may find some other way to stabilize itself. In such cases, Y never attacks at all and the product is cyclic. These are simple internal SN2 reactions.117 Two examples are formation of epoxides and lactones:
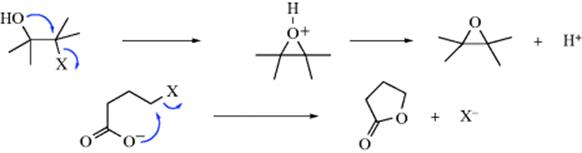
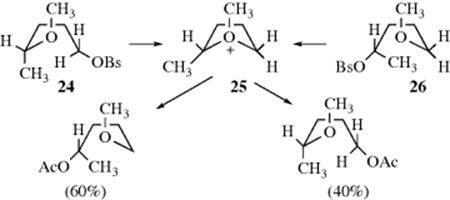
The fact that acetolysis of both 4-methoxy-1-pentyl brosylate (24) and 5-methoxy- 2-pentyl brosylate (25) gave the same mixture of products is further evidence for participation by a neighboring group.118 In this case, the intermediate 26 is common to both substrates.
The neighboring-group mechanism operates only when the ring size is right for a particular type of Z. For example, for MeO(CH2)nOBs, neighboring-group participation was important for n = 4 or 5 (corresponding to a five- or six-membered intermediate), but not for n = 2, 3, or 6.119 However, optimum ring size is not the same for all reactions, even with a particular Z. In general, the most rapid reactions occur when the ring size is three, five, or six, depending on the reaction type. The likelihood of four-membered ring neighboring-group participation is increased when there are alkyl groups α or β to the neighboring group.120
The following are some of the more important neighboring groups: COO− (but not COOH), COOR, COAr, OCOR,121 OR, OH, O−,122 NH2, NHR, NR2, NHCOR, SH, SR, S−,123 SO2Ph,124 I, Br, and Cl. The effectiveness of halogens as neighboring groups decreases in the order I > Br > Cl.125 The Cl substituents is a very weak neighboring group, and can be shown to act in this way only when the solvent does not interfere. For example, when 5-chloro-2-hexyl tosylate is solvolyzed in acetic acid, there is little participation by the Cl, but when the solvent is changed to trifluoroacetic acid, which is much less nucleophilic, neighboring-group participation by the Cl becomes the major reaction pathway.126 Thus, Cl acts as a neighboring group only when there is need for it (for other examples of the principle of increasing electron demand, see Sec. 10.C.i).
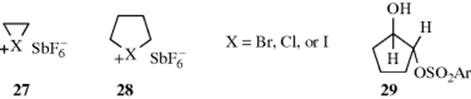
A number of intermediates of halogen participation (halonium ions),127 (e.g., 27 and 28), have been prepared as stable salts in SbF5–SO2 or SbF5–SO2ClF solutions.128 Some have even been crystallized. Attempts to prepare four-membered homologues of 27 and 28 were not successful.129 There is no evidence that F can act as a neighboring group.123
The principle that a neighboring group lends assistance in proportion to the need for such assistance also applies to differences in leaving-group ability. Thus, p-NO2C6H4SO2O (the nosylate group) is a better leaving group than p-MeC6H4SO2O (the tosylate group). Experiments have shown that the OH group in trans-2-hydroxycyclopentyl arenesulfonates (29) acts as a neighboring group when the leaving group is tosylate, but not when it is nosylate, apparently because the nosylate group leaves so rapidly that it does not require assistance.130
10.C.i. Neighboring-Group Participation by π and σ Bonds: Nonclassical Carbocations131
For all the neighboring groups listed in Section 10.C, the nucleophilic attack is made by an atom with an unshared pair of electrons. In this section, neighboring-group participation by C=C π bonds and C–C and C–H σ bonds will be considered. There has been a great deal of controversy over whether such bonds can act as neighboring groups and about the existence and structure of the intermediates involved. These intermediates are called nonclassical (or bridged) carbocations. In classical carbocations (Chap 5), the positive charge is localized on one carbon atom or delocalized by resonance involving an unshared pair of electrons or a double or triple bond in the allylic position. In a nonclassical carbocation,132 the positive charge is delocalized by a double or triple bond that is not in the allylic position or by a single bond. Examples are the 7-norbornenyl cation (30), the norbornyl cation (31),133 and the cyclopropylmethyl cation (32). A cyclopropyl group (as in 33) is capable of stabilizing the norbornyl cation, inhibiting this rearrangement.134 Carbocation 30 is called a homoallylic carbocation, because in 30a there is one carbon atom between the positively charged carbon and the double bond. Many of these carbocations can be produced in more than one way if the proper substrates are chosen. For example, 31 can be generated by the departure of a leaving group from 34 or from 35.135 The first of these pathways is called the σ route to a nonclassical carbocation, because participation of a σ bond is involved. The second is called the π route.136 The argument against the existence of nonclassical carbocations is essentially that the structures 30a–30c (or 31a, 31b, etc.) are not canonical forms, but real structures and that there is rapid equilibration among them. This debate remained an active area of interest for some reactions for many years.137 In one study, the solvolysis and rearrangement of 2-bicyclo[3.2.2]nonanyl tosylate in methanol generated ethers derived from the 2-bicyclo[3.2.2]-nonanyl and 2-bicyclo[3.3.1]nonanyl systems that were rationalized in terms of a classical carbocation.138 Density functional and ab initio calculations indicated that the products of the 2-bicyclo[3.2.2]nonanyl tosylate solvolysis were found to have nonclassical structures.139
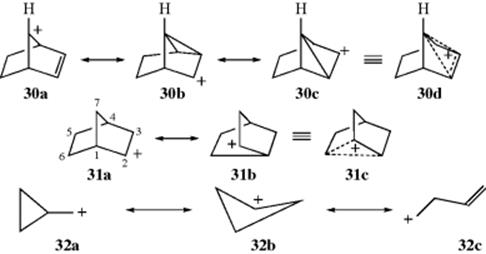
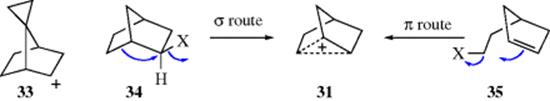
In discussing nonclassical carbocations, care must be taken to make the distinction between neighboring-group participation and the existence of nonclassical carbocations.140 If a nonclassical carbocation exists in any reaction, then an ion with electron delocalization, as shown in the above examples, is a discrete reaction intermediate. If a carbon–carbon double or single bond participates in the departure of the leaving group to form a carbocation, it may be that a nonclassical carbocation is involved, but there is no necessary relation. In any particular case, either or both of these possibilities are possible.
In the following pages, some of the evidence bearing on the questions of the participation of π and σ bonds is considered, which bears on the existence of nonclassical carbocations,141 although a thorough discussion is beyond the scope of this book.100
1. C=C as a Neighboring Group.142 The most striking evidence that C=C can act as a neighboring group is that acetolysis of 36–OTs is 1011 times faster than that of 37–OTs and proceeds with retention of configuration.143 The rate data alone do not necessarily prove that acetolysis of 36–OTs involves a nonclassical intermediate (30d), but it is certainly strong evidence that the C=C group assists in the departure of the OTs.
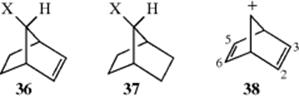
Evidence that 30 is indeed a nonclassical ion comes from an NMR study of the relatively stable norbornadienyl cation (38). The ![]() NMR spectrum shows that the 2 and 3 protons are not equivalent to the 5 and 6 protons.144Thus there is interaction between the charged carbon and one double bond, which is evidence for the existence of 30d.145 In the case of 36, the double bond is geometrically fixed in an especially favorable position for backside attack on the carbon bearing the leaving group (hence the very large rate enhancement), but there is much evidence that other double bonds in the homoallylic position,146 as well as in positions farther away,147 can also lend anchimeric assistance, although generally with much lower rate ratios. One example of the latter is the compound β-(syn-7-norbornenyl)ethyl brosylate (39), which at 25°C undergoes acetolysis ~ 140,000 times faster than the saturated analogue 40.148 Triple bonds149 and allenes150 can also act as neighboring groups.
NMR spectrum shows that the 2 and 3 protons are not equivalent to the 5 and 6 protons.144Thus there is interaction between the charged carbon and one double bond, which is evidence for the existence of 30d.145 In the case of 36, the double bond is geometrically fixed in an especially favorable position for backside attack on the carbon bearing the leaving group (hence the very large rate enhancement), but there is much evidence that other double bonds in the homoallylic position,146 as well as in positions farther away,147 can also lend anchimeric assistance, although generally with much lower rate ratios. One example of the latter is the compound β-(syn-7-norbornenyl)ethyl brosylate (39), which at 25°C undergoes acetolysis ~ 140,000 times faster than the saturated analogue 40.148 Triple bonds149 and allenes150 can also act as neighboring groups.
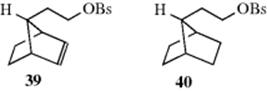
Evidence has been presented to show that participation by a potential neighboring group can be reduced or eliminated if an outside nucleophile is present that is more effective than the neighboring group in attacking the central carbon (see above), or if a sufficiently good leaving group is present (see above). In another example of the principle of increasing electron demand, Gassman et al.151 showed that neighboring-group participation can also be reduced if the stability of the potential carbocation is increased. They found that the presence of a p-anisyl group at the 7 position of 36 and 37 exerts a powerful leveling effect on the rate differences. Thus, solvolysis in acetone–water at 85 °C of 38 was only ~ 2.5 times faster than that of the saturated compound 42. Furthermore, both 41 and its stereoisomer 43 gave the same mixture of solvolysis products, showing that the stereoselectivity in the solvolysis of 36 is not present here. The difference between 41 and 36 is that in the case of 41 the positive charge generated at the 7 position in the transition state is greatly stabilized by the p-anisyl group. Apparently, the stabilization by the p-anisyl group is so great that further stabilization that would come from participation by the C=C bond is not needed.152 The use of a phenyl instead of a p-anisyl group is not sufficient to stop participation by the double bond completely, although it does reduce it.153 These results essentially emphasize the previous conclusion that a neighboring group lends anchimeric assistance only when there is sufficient demand for it.154 The π-bond of a neighboring alkene group can assist solvolysis via π-participation.155
The ability of C=C to serve as a neighboring group can depend on its electron density. When the strongly electron-withdrawing CF3 group was attached to a double-bond carbon of 44, the solvolysis rate was lowered by a factor of ~ 106.156 A second CF3 group had an equally strong effect. In this case, two CF3 groups decrease the electron density of the C=C bond to the point that the solvolysis rate for 44 (R1 = R2 = CF3) was about the same as (actually ~ 17 times slower than) the rate for the saturated substrate 37 (X = OMos). Thus, the two CF3 groups completely remove the ability of the C=C bond to act as a neighboring group.
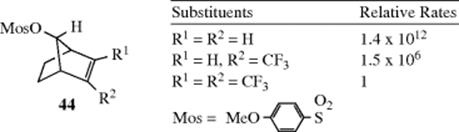
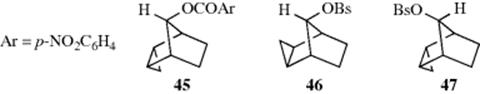
2. Cyclopropyl157as a Neighboring Group.158 In Section 4.Q.i, the properties of a cyclopropane ring were shown to be similar to those of a double bond in some ways. Therefore it is not surprising that a suitably placed cyclopropyl ring can also be a neighboring group. Thus endo–anti-tricyclo[3.2.1.02,4]octan-8-yl p-nitrobenzoate (45) solvolyzed ~ 1014 times faster that the p-nitrobenzoate of 37–OH.159 Obviously, a suitably placed cyclopropyl ring can be even more effective160 as a neighboring group than a double bond.161 The need for suitable placement is emphasized by the fact that 47 solvolyzed only about five times faster than 37–OBs,162 while 46solvolyzed three times slower than 37–OBs.163 In the case of 45 and of all other cases known where cyclopropyl lends considerable anchimeric assistance, the developing p orbital of the carbocation is orthogonal to the participating bond of the cyclopropane ring.164 An experiment designed to test whether a developing p orbital that would be parallel to the participating bond would be assisted by that bond showed no rate enhancement.164 This result is in contrast to the behavior of cyclopropane rings directly attached to positively charged carbons, where the p orbital is parallel to the plane of the ring (Sec. 5.A.ii, and category 4.b below). Rate enhancements, although considerably smaller, have also been reported for suitably placed cyclobutyl rings.165
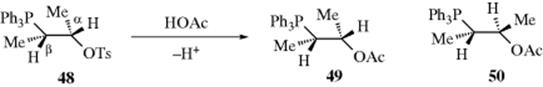
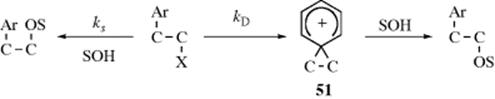
3. Aromatic Rings as Neighboring Groups.166 There is a great deal of evidence that aromatic rings in the β position can function as neighboring groups.167 Stereochemical evidence was obtained by solvolysis of L-threo-3-phenyl-2-butyl tosylate (48) in acetic acid.168 Of the acetate product, 96% was the threo isomer and only ~ 4% was erythro. Moreover, both the (+) and (-) threo isomers (49 and 50) were produced in approximately equal amounts (a racemic mixture). When solvolysis was conducted in formic acid, even less erythro isomer was obtained. This result is similar to that found on reaction of 3-bromo-2-butanol with HBr (Sec. 10.C) and leads to the conclusion that configuration is retained because phenyl acts as a neighboring group. However, evidence from rate studies is not so simple. If β-aryl groups assist the departure of the leaving group, solvolysis rates should be enhanced. In general, they are not. However, solvolysis rate studies in 2-arylethyl systems are complicated by the fact that, for primary and secondary systems, two pathways can exist.169 In one of these (designated kΔ), the aryl, behaving as a neighboring group, pushes out the leaving group to give a bridged ion, called a phenonium ion (51), and is in turn pushed out by the solvent (SOH), so the net result is substitution with retention of configuration (or rearrangement, if 51 is opened from the other side). The other pathway (ks) is simple SN2 attack by the solvent at the leaving-group carbon. The net result is substitution with inversion and no possibility of rearrangement. Whether the leaving group is located at a primary or a secondary carbon, there is no cross over between these pathways; they are completely independent.170 Both the kΔ and ks pathways are unimportant when the leaving group is at a tertiary carbon. In these cases, the mechanism is SN1 and open carbocations ArCH2CR2+ are intermediates. This pathway is designated kc. Which of the two pathways (ks or kΔ) predominates in any given case depends on the solvent and on the nature of the aryl group. As expected from the results we have seen for Cl as a neighboring group (see above), the kΔ/ks ratio is highest for solvents that are poor nucleophiles and so compete very poorly with the aryl group. For several common solvents, the kΔ/ks ratio increases in the order EtOH < CH3CO2H < HCO2H < CF3CO2H.171 In accord with this, the following percentages of retention were obtained in solvolysis of 1-phenyl-2-propyl tosylate at 50 °C: solvolysis in EtOH 7%, CH3CO2H 35%, and HCO2H 85%.171 This finding indicates that ks predominates in EtOH (phenyl participates very little), while kΔ predominates in HCO2H. Trifluoroacetic acid is a solvent of particularly low nucleophilic power, and in this solvent the reaction proceeds entirely by kΔ;172 deuterium labeling showed 100% retention.173 This case provides a clear example of neighboring-group rate enhancement by phenyl. The rate of solvolysis of PhCH2CH2OTs at 75 °C in CF3COOH is 3040 times the rate for CH3CH2OTs.172
With respect to the aromatic ring, the kΔ pathway is electrophilic aromatic substitution (Chapter 11). Groups on the ring that activate that reaction (Sec. 11.B.i) are predicted to increase, and deactivating groups will decrease the rate of this pathway. This prediction has been borne out by several investigations. The p-nitro derivative of 48 solvolyzed in acetic acid 190 times slower than 48, and there was much less retention of configuration; the acetate produced was only 7% threo and 93% erythro.174 At 90 °C, acetolysis of p-ZC6H4CH2CH2OTs gave the rate ratios shown in Table 10.1.175 Throughout this series ks is fairly constant, as it should be since it is affected only by the rather remote field effect of Z. It is kΔ that changes substantially as Z is changed from activating to deactivating. The evidence is thus fairly clear that participation by aryl groups depends greatly on the nature of the group. For some groups (e.g., p-nitrophenyl), in some solvents (e.g., acetic acid), there is essentially no neighboring-group participation at all,176 while for others (e.g., p-methoxyphenyl),neighboring-group participation is substantial. The combined effect of solvent and structure is shown in Table 10.2, where the figures shown were derived by three different methods.177 The decrease in neighboring-group effectiveness when aromatic rings are substituted by electron-withdrawing groups is reminiscent of the similar case of C=C bonds substituted by CF3 groups (see above, category 1).
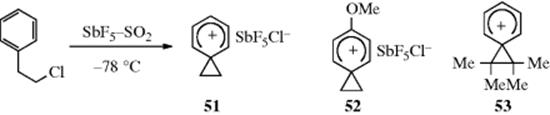
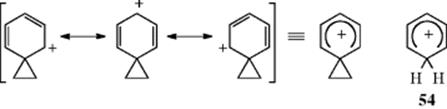
Several phenonium ions have been prepared as stable ions in solution where they can be studied by NMR, among them 52,17853,179and the unsubstituted 51.180 These were prepared181 by the method shown for 51: treatment of the corresponding β-arylethyl chloride with SbF5–SO2 at low temperatures. These conditions are even more extreme than the solvolysis in CF3COOH mentioned earlier. The absence of any nucleophile eliminates not only the ks pathways but also nucleophilic attack on 51. Although 51 is not in equilibrium with the open-chain ion PhCH2CH2+, which is primary and hence unstable, 53 is in equilibrium with the open-chain tertiary ions PhCMe2C+Me2and PhC+MeCMe3, although only 53 is present in appreciable concentration. Proton and ![]() NMR show that 51–53 are classical carbocations where the only resonance is in the six-membered ring. The three-membered ring is a normal cyclopropane ring that is influenced only to a relatively small extent by the positive charge on the adjacent ring. Nuclear magnetic resonance spectra show that the six-membered rings have no aromatic character, but are similar in structure to the arenium ions (e.g., 54), that are intermediates in electrophilic aromatic substitution (Chapter 11). A number of phenonium ions, including 51, have also been reported to be present in the gas phase, where their existence has been inferred from reaction products and from
NMR show that 51–53 are classical carbocations where the only resonance is in the six-membered ring. The three-membered ring is a normal cyclopropane ring that is influenced only to a relatively small extent by the positive charge on the adjacent ring. Nuclear magnetic resonance spectra show that the six-membered rings have no aromatic character, but are similar in structure to the arenium ions (e.g., 54), that are intermediates in electrophilic aromatic substitution (Chapter 11). A number of phenonium ions, including 51, have also been reported to be present in the gas phase, where their existence has been inferred from reaction products and from ![]() labeling.182
labeling.182
It is thus clear that β-aryl groups can function as neighboring groups.183 Much less work has been done on aryl groups located in positions farther away from the leaving group, but there is evidence that these too can lend anchimeric assistance.184
4. The Carbon–Carbon Single Bond as a Neighboring Group.185
a. The 2-Norbornyl System. In the investigations to determine whether a C–C σ bond can act as a neighboring group, by far the greatest attention has been paid to the 2-norbornyl system.186 Winstein et al.187 found that solvolysis in acetic acid of optically active exo-2-norbornyl brosylate (55, OB = brosylate) gave a racemic mixture of the two exo acetates; no endo isomers were formed:
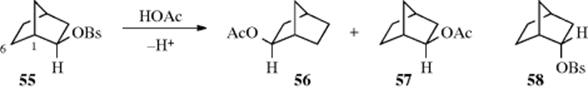
Furthermore, 55 solvolyzed ~ 350 times faster than its endo isomer (58). Similar high exo/endo rate ratios have been found in many other [2.2.1] systems. These two results [(1) that solvolysis of an optically active exo isomer gave only racemic exo isomers and (2) the high exo/endo rate ratio] were interpreted by Winstein et al.187 as indicating that the 1,6-bond assists in the departure of the leaving group and that a nonclassical intermediate (59) is involved. They reasoned that solvolysis of the endo isomer (58) is not assisted by the 1,6-bond because it is not in a favorable position for backside attack, and that consequently solvolysis of 58 takes place at a “normal” rate. Therefore the much faster rate for the solvolysis of 55 must be caused by anchimeric assistance. The stereochemistry of the product is also explained by the intermediacy of 59, since in 59 the 1 and 2 positions are equivalent and would be attacked by the nucleophile with equal facility, but only from the exo direction in either case. Incidentally, acetolysis of 58 also leads exclusively to the exo acetates (56 and 57), so that in this case Winstein et al.187 postulated that a classical ion (60) is first formed and then converted to the more stable 59. Evidence for this interpretation is that the product from solvolysis of 58 is not racemic, but contains somewhat more 57 than 56 (corresponding to 3–13% inversion, depending on the solvent), suggesting that when 60 is formed, some of it goes to give 57 before it can collapse to 59.
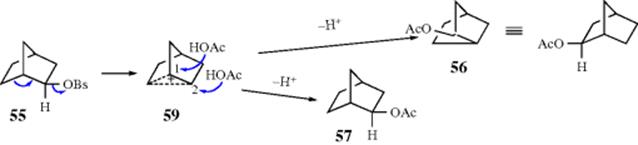
The concepts of σ participation and the nonclassical ion 59 were challenged by H.C. Brown,141 who suggested that the two results can also be explained by postulating that 55 solvolyzes without participation of the 1,6 bond to give the classical ion 60, which is in rapid equilibrium with 61. This rapid interconversion has been likened to the action of a windshield wiper.188 Obviously, in going from 60 to 61 and back again, 59 must be present, but in Brown's view it is a transition state and not an intermediate. Brown's explanation for the stereochemical result was that exclusive exo attack is a property to be expected from any 2-norbornyl system, not only for the cation but even for reactions not involving cations, because of steric hindrance to attack from the endo side. There is a large body of data showing that exo attack on norbornyl systems is fairly general in many reactions. A racemic mixture will be obtained if 60 and 61 are present in equal amounts, since they are equivalent and exo attack on 60 and 61 gives, respectively, 57 and 56. Brown explained the high exo/endo rate ratios by contending that it is not the endo rate that is normal and the exo rate abnormally high, but the exo rate that is normal and the endo rate abnormally low, because of steric hindrance to removal of the leaving group in that direction.189
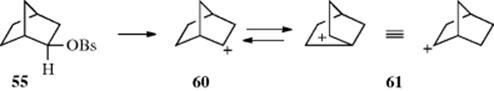
A vast amount of work has been done190 on solvolysis of the 2-norbornyl system in an effort to determine whether the 1,6-bond participates and whether 59 is an intermediate. Most,191 although not all,192 chemists now accept the intermediacy of 59.
Besides the work done on solvolysis, of 2-norbornyl compounds, the 2-norbornyl cation has also been extensively studied at low temperatures; there is much evidence that under these conditions the ion is definitely nonclassical. Olah and co-workers prepared the 2-norbornyl cation in stable solutions at temperatures below -150 °C in SbF5-SO2 and FSO3H–SbF5–SO2, where the structure is static and hydride shifts are absent.193,194Studies by ![]() and
and ![]() NMR, as well as by laser Raman spectra and X-ray electron spectroscopy, led to the conclusion194 that under these conditions the ion is nonclassical.195 A similar result has been reported for the 2-norbornyl cation in the solid state, where at 77 and even 5 K,
NMR, as well as by laser Raman spectra and X-ray electron spectroscopy, led to the conclusion194 that under these conditions the ion is nonclassical.195 A similar result has been reported for the 2-norbornyl cation in the solid state, where at 77 and even 5 K, ![]() NMR spectra gave no evidence of the freezing out of a single classical ion.196
NMR spectra gave no evidence of the freezing out of a single classical ion.196
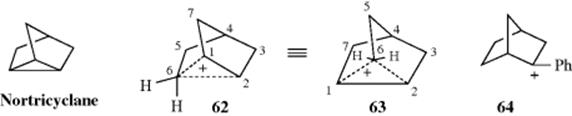
Olah and co-workers represented the nonclassical structure as a corner-protonated nortricyclane (62); the symmetry is better seen when the ion is drawn, as in 63. Almost all the positive charge resides on C-1 and C-2 and very little on the bridging carbon C-6. Other evidence for the nonclassical nature of the 2-norbornyl cation in stable solutions comes from heat of reaction measurements showing that the 2-norbornyl cation is more stable (by ~ 6–10 kcal mol−1 or 25–40 kJ mol−1) than would be expected without the bridging.197 Studies of IR spectra of the 2-norbornyl cation in the gas phase also show the nonclassical structure.198Ab initio calculations show that the nonclassical structure corresponds to an energy minimum.199
The spectra of other norbornyl cations have also been investigated at low temperatures. Spectra of the tertiary 2-methyl- and 2-ethylnorbornyl cations show less delocalization,200 and the 2-phenylnorbornyl cation (64) is essentially classical,201 as are the 2-methoxy-202 and 2-chloronorbornyl cations.203 Recall (Sec. 5.A.ii) that methoxy and halo groups also stabilize a positive charge. The ![]() NMR data show that electron-withdrawing groups on the benzene ring of 64 cause the ion to become less classical, while electron-donating groups enhance the classical nature of the ion.204
NMR data show that electron-withdrawing groups on the benzene ring of 64 cause the ion to become less classical, while electron-donating groups enhance the classical nature of the ion.204
b. The Cyclopropylmethyl System. Apart from the 2-norbornyl system, the greatest amount of effort in the search for C–C participation has been devoted to the cyclopropylmethyl system.205 It has long been known that cyclopropylmethyl substrates solvolyze with abnormally high rates and that the products often include not only unrearranged cyclopropylmethyl, but also cyclobutyl and homoallylic compounds. An example is206
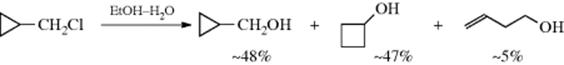
Cyclobutyl substrates also solvolyze abnormally rapidly and give similar products. Indeed, computational studies on the cyclobutylmethyl cation suggest it is nonclassical (Sec. 10.C.i). Furthermore, when the reactions are carried out with labeled substrates, considerable, although not complete, scrambling is observed. For these reasons, it has been suggested that a common intermediate (some kind of nonclassical intermediate, e.g., 32) is present in these cases. This common intermediate could then be obtained by three routes:
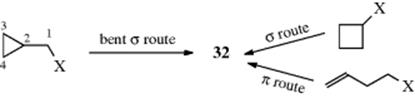
In recent years, much work has been devoted to the study of these systems, and it is apparent that matters are not so simple. although there is much that is still not completely understood, some conclusions can be drawn.
i. In the solvolysis of simple primary cyclopropylmethyl systems, the rate is enhanced because of participation by the σ bonds of the ring.207 The ion that forms initially is an unrearranged cyclopropylmethyl cation208that is symmetrically stabilized; that is, both the 2,3 and 2,4 σ bonds help stabilize the positive charge. As seen previously (Sec. 5.A.ii) that a cyclopropyl group stabilizes an adjacent positive charge even better than a phenyl group. One way of representing the structure of this cation is as shown in 65. Among the evidence that 65 is a symmetrical ion is that substitution of one or more methyl groups in the 3 and 4 positions increases the rate of solvolysis of cyclopropylcarbinyl 3,5-dinitrobenzoates by approximately a factor of 10 for each methyl group.209 If only one of the σ bonds (say, the 2,3 bond) stabilizes the cation, then methyl substitution at the 3 position should increase the rate, and a second methyl group at the 3 position should increase it still more, but a second methyl group at the 4 position should have little effect.210
![]()
ii. The most stable geometry of simple cyclopropylmethyl cations is the bisected one shown in Section 5.A.ii. There is much evidence that in systems where this geometry cannot be obtained, solvolysis is greatly slowed.211
iii. Once a cyclopropylmethyl cation is formed, it can rearrange to two other cyclopropylmethyl cations:
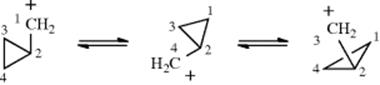
This rearrangement, which accounts for the scrambling, is completely stereospecific.212 The rearrangements probably take place through a nonplanar cyclobutyl cation intermediate or transition state. The formation of cyclobutyl and homoallylic products from a cyclopropylmethyl cation is also completely stereospecific. These products may arise by direct attack of the nucleophile on 65 or on the cyclobutyl cation intermediate.213 A planar cyclobutyl cation is ruled out in both cases because it would be symmetrical and the stereospecificity would be lost.
iv. The rate enhancement in the solvolysis of secondary cyclobutyl substrates is probably caused by participation by a bond leading directly to 65, which accounts for the fact that solvolysis of cyclobutyl and of cyclopropylmethyl substrates often gives similar product mixtures. There is no evidence that requires cyclobutyl cations to be intermediates in most secondary cyclobutyl systems, although tertiary cyclobutyl cations can be solvolysis intermediates.
![]()
v. The unsubstituted cyclopropylmethyl cation has been generated in superacid solutions at low temperatures, where ![]() NMR spectra have led to the conclusion that it consists of a mixture of the bicyclobutonium ion (32) and the bisected cyclopropylmethyl cation (65), in equilibrium with 32.214 Molecular orbital calculations show that these two species are energy minima, and that both have nearly the same energy.213
NMR spectra have led to the conclusion that it consists of a mixture of the bicyclobutonium ion (32) and the bisected cyclopropylmethyl cation (65), in equilibrium with 32.214 Molecular orbital calculations show that these two species are energy minima, and that both have nearly the same energy.213
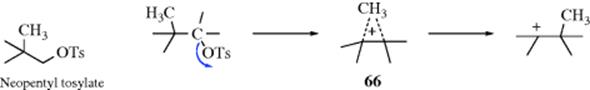
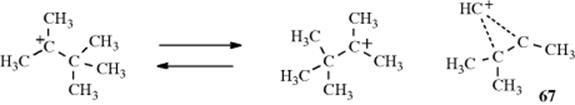
c. Methyl as a Neighboring Group. Both the 2-norbornyl and cyclopropylmethyl system contain a σ bond that is geometrically constrained to be in a particularly favorable position for participation as a neighboring group. However, there have been a number of investigations to determine whether a C–C bond can lend anchimeric assistance even in a simple open-chain compound, (e.g., neopentyl tosylate). On solvolysis, neopentyl systems undergo almost exclusive rearrangement and 66 must lie on the reaction path, but the two questions that have been asked are (1) Is the departure of the leaving group concerted with the formation of the CH3–C bond (e.g., does the methyl participate)? (2) Is 66 an intermediate or only a transition state? With respect to the first question, there is evidence, chiefly from isotope effect studies, which indicates that the methyl group in the neopentyl system does indeed participate,215 although it may not greatly enhance the rate. As to the second question, evidence that 66 is an intermediate is that small amounts of cyclopropanes (10–15%) can be isolated in these reactions.216 Cation 66 is a protonated cyclopropane and would give cyclopropane on loss of a proton.217 In an effort to isolate a species that has structure 66, the 2,3,3-trimethyl-2-butyl cation was prepared in superacid solutions at low temperatures.218 However, ![]() and
and ![]() NMR, as well as Raman spectra, showed this to be a pair of rapidly equilibrating open ions. Of course, 67 must lie on the reaction path connecting the two open ions, but it is evidently a transition state and not an intermediate. However, evidence from X-ray photoelectron spectroscopy (XPS) has shown that the 2-butyl cation is substantially methyl bridged.219
NMR, as well as Raman spectra, showed this to be a pair of rapidly equilibrating open ions. Of course, 67 must lie on the reaction path connecting the two open ions, but it is evidently a transition state and not an intermediate. However, evidence from X-ray photoelectron spectroscopy (XPS) has shown that the 2-butyl cation is substantially methyl bridged.219
d. Silylalkyl as a Neighboring Group. Rates of solvolysis are enhanced in molecules that contain a silylalkyl or silylaryl group β-to the carbon bearing the leaving group. This is attributed to formation of a cyclic transition state involving the silicon.220
5. Hydrogen as a Neighboring Group. The questions relating to hydrogen are similar to those relating to methyl. There is no question that hydride can migrate, but the two questions are (1) Does the hydrogen atom participate in the departure of the leaving group? (2) Is 68 an intermediate or only a transition state? There is some evidence that a β hydrogen can participate.221 Evidence that 68 can be an intermediate in solvolysis reactions comes from a study of the solvolysis in trifluoroacetic acid of deuterated sec-butyl tosylate 69. In this solvent of very low nucleophilic power, the products were an equimolar mixture of 70 and 71,222 but
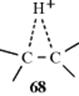
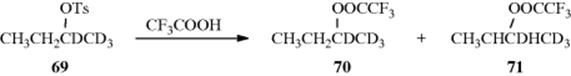
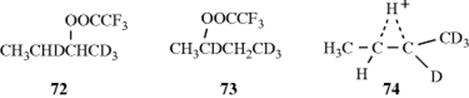
no72 or 73 was found. If this reaction did not involve neighboring hydrogen at all (pure SN2 or SN1), the product would be only 70. On the other hand, if hydrogen does migrate, but only open cations are involved, then there should be an equilibrium among these four cations:
![]()
leading not only to 70 and 71, but also to 72 and 73. The results are most easily compatible with the intermediacy of the bridged ion 74, which can then be attacked by the solvent equally at the 2 and 3 positions. Attempts to prepare 68 as a stable ion in superacid solutions at low temperatures have not been successful.221
Table 10.1 Approximate kΔ/ks Ratios for Acetolysis of p-ZC6H4CH2CH2OTs at 90 °Ca
Z
kΔ/ks
MeO
30
Me
11
H
1.3
Cl
0.3
Reprinted with permission Jones, M.G.; Coke, J.L. J. Am. Chem. Soc. 1969 , 91, 4284. Copyright © 1969 American Chemical Society.
a. See Ref. 145.
Table 10.2 Percent of Product Formed by the kΔ Pathway in Solvolysis of p-ZC6H4CH2CH2OTsa
Z
Solvent
Percent by kΔ
H
CH3COOH
35–38
H
HCOOH
72–79
MeO
CH3COOH
91–93
MeO
HCOOH
99
Reprinted with permission Lancelot, C.J.; Schleyer, P.v.R. J. Am. Chem. Soc. 1969, 91, 4296. Copyright © 1969 American Chemical Society.
a. See Ref. 177.