March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 10. Aliphatic Substitution, Nucleophilic and Organometallic
10.G. Reactivity
A large amount of work has been done on this subject. Although a great deal is known, much is still poorly understood, and many results are anomalous and hard to explain. In this section, only approximate generalizations are attempted. The work discussed here, and the conclusions reached, pertain to reactions taking place in solution. Some investigations have also been carried out in the gas phase.282
10.G.i. The Effect of Substrate Structure
The effect on the reactivity of a change in substrate structure depends on the mechanism.
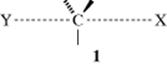
1. Branching at the α and β Carbons. For the SN2 mechanism, branching at either the α or the β carbon decreases the rate. Tertiary systems seldom283 react by the SN2 mechanism and neopentyl systems react so slowly as to make such reactions, in general, synthetically useless.284 Experiments show that methyl halide reacts 30 times faster than ethyl, whereas isopropyl halides react 40 times slower than ethyl.285 The presence of π bonds accelerates the rate, as illustrated by the fact that allyl halides react 40 times faster than ethyl halides, and benzyl halides react 120 times faster.285 The reason for the low rates for secondary and especially tertiary is almost certainly steric. This statement includes the primary neopentyl halides, which react 20,000 times slower than ethyl halides286 The transition state 1 is more crowded when larger groups are close to the central carbon.287
The tetrahedral mechanism for substitution at a carbonyl carbon (Chapter 16) is also slowed or blocked completely by α or β branching for similar reasons. Solvolysis in such systems is linked to relief of B-strain, but solvent participation can overshadow this as steric hindrance increases.288 Severe steric strain can cause distortion from coplanarity in the carbocation intermediate,289 although there seems to be no loss of resonance stability.290 Adding electron-donating substituents to such molecules improves coplanarity in the cation.291 For example, esters of the formula R3CCOOR′ cannot generally be hydrolyzed by the tetrahedral mechanism (see Reaction 16-59), and R3CCO2H acids cannot be easily esterified.292 Synthetic advantage can be taken of this fact, for example, with a molecule containing two ester groups only the less hindered one is hydrolyzed.
For the SN1 mechanism, branching as the α carbon increases the rate, as shown by rate data for alkyl bromides.293 The secondary bromide isopropyl bromide reacts 11.6 times faster than bromoethane in water at 50 °C, and tert-butyle bromide (a tertiary halide) reacts 1.2 × 106 times faster293. This result is explained by the stability order of alkyl cations (tertiary > secondary > primary). Of course, the rates are not actually dependent on the stability of the ions, but on the difference in free energy between the starting compounds and the transition states. The Hammond postulate (Sec. 6.G) is used to make the assumption that the transition states resemble the cations and that anything (e.g., branching) that lowers the free energy of the ions also lowers it for the transition states. For simple alkyl groups, the SN1 mechanism is important under all conditions only for tertiary substrates.294 As previously indicated (Sec. 10.A.iv), secondary substrates generally react by the SN2 mechanism,295 except that the SN1 mechanism may become important at high solvent polarities. Isopropyl bromide reacts less than twice as fast as ethyl bromide in the relatively nonpolar 60% ethanol (cf. this with the 104 ratio for tert-butylbromide, where the mechanism is certainly SN1), but in the more polar water the rate ratio is 11.6.293 The 2-adamantyl system is an exception; it is a secondary system that reacts by the SN1 mechanism because backside attack is hindered for steric reasons.296 Because there is no SN2 component, this system provides an opportunity for comparing the pure SN1 reactivity of secondary and tertiary substrates. It has been found that substitution of a methyl group for a hydrogen of 2-adamantyl substrates (thus changing a secondary to a tertiary system) increases solvolysis rates by a factor of ~ 108.297 Simple primary substrates react by the SN2 mechanism (or with participation by neighboring alkyl or hydrogen), but not by the SN1 mechanism, even when solvolyzed in solvents of very low nucleophilicity298 (e.g., trifluoroacetic acid or trifluoroethanol299), and even when very good leaving groups (e.g., OSO2F) are present300 (see, however, Sec. 10.G.iii).
For some tertiary substrates, the rate of SN1 reactions is greatly increased by the relief of B strain in the formation of the carbocation (see Sec. 9.B). Except where B strain is involved, β branching has little effect on the SN1 mechanism, except that carbocations with β branching undergo rearrangements readily. Of course, isobutyl and neopentyl are primary substrates, and for this reason react very slowly by the SN1 mechanism, but not more slowly than the corresponding ethyl or propyl compounds.
To sum up, primary and secondary substrates generally react by the SN2 mechanism and tertiary by the SN1 mechanism. However, tertiary substrates seldom undergo nucleophilic substitution at all. Elimination is always a possible side reaction of nucleophilic substitutions (wherever a β hydrogen is present), and with tertiary substrates it usually predominates. With a few exceptions, nucleophilic substitutions at a tertiary carbon have little or no preparative value. However, tertiary substrates that can react by the SET mechanism (e.g., p-NO2C6H4CMe2Cl) give very good yields of substitution products when treated with a variety of nucleophiles.301
2. Unsaturation at the α Carbon. Vinylic, acetylenic,302 and aryl substrates are very unreactive toward nucleophilic substitutions. For these systems, both the SN1 and SN2 mechanisms are greatly slowed or stopped altogether. One reason that has been suggested for this is that sp2 (and even more, sp) carbon atoms have a higher electronegativity than sp3 carbons and thus a greater attraction for the electrons of the bond. As seen previously (Sec. 8.F, category 7), an sp-H bond has a higher acidity than an sp3-H bond, with that of an sp2–H bond in between. This is reasonable; the carbon retains the electrons when the proton is lost and an sp carbon, which has the greatest hold on the electrons, loses the proton most easily. But in nucleophilic substitution, the leaving group carries off the electron pair, so the situation is reversed and it is the sp3 carbon that loses the leaving group and the electron pair most easily. Recall (Sec. 1.J) that bond distances decrease with increasing s character. Thus the bond length for a vinylic or aryl C–Cl bond is 1.73 Å compared with 1.78 Å for a saturated C–Cl bond. Other things being equal, a shorter bond is a stronger bond.
Of course, it has been seen (Sec. 10.F) that SN1 reactions at vinylic substrates can be accelerated by α substituents that stabilize that cation, and that reactions by the tetrahedral mechanism can be accelerated by β substituents that stabilize the carbanion. Also, reactions at vinylic substrates can in certain cases proceed by addition–elimination or elimination–addition sequences (Sec. 10.F).In contrast to such systems, substrates of the type RCOX are usually much more reactive than the corresponding RCH2X. Of course, the mechanism here is almost always the tetrahedral one. Three reasons can be given for the enhanced reactivity of RCOX: (1) The carbonyl carbon has a sizable partial positive charge that makes it very attractive to nucleophiles. (2) In an SN2 reaction, a σ bond must break in the rate-determining step, which requires more energy than the shift of a pair of π electrons, which is what happens in a tetrahedral mechanism. (3) A trigonal carbon offers less steric hindrance to a nucleophile than a tetrahedral carbon.
For reactivity in aryl systems, see Chapter 13.
3. Unsaturation at the β Carbon. The SN1 rates are increased when there is a double bond in the β position, so that allylic and benzylic substrates react rapidly (allylic tosylates react more than 30 times faster than ethyl tosylate).303 The reason is that allylic (Sec. 5.A.ii) and benzylic304 (Sec. 5.A.ii) cations are stabilized by resonance. The presence of a second or a third phynyl group increases the rate still more (105 and 1010 times faster, respectively), because these carbocations are more stable yet.303 Remember that allylic rearrangements are possible with allylic systems.
In general, SN1 rates at an allylic substrate are increased by any substituent in the 1 or 3 position that can stabilize the carbocation by resonance or hyperconjugation.305 Among these are alkyl, aryl, and halo groups.
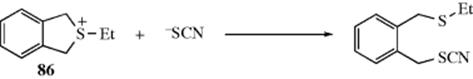
The SN2 rates for allylic and benzylic systems are also increased (see above), probably owing to resonance possibilities in the transition state. Evidence for this in benzylic systems is that the rate of the reaction was 8000 times slower than the rate with (PhCH2)2SEt+.306 The cyclic 86 does not have the proper geometry for conjugation in the transition state.
Triple bonds in the β position (in propargyl systems) have about the same effect as double bonds.307 Alkyl, aryl, halo, and cyano groups, among others, in the 3 position of allylic substrates increase SN2 rates, owing to increased resonance in the transition state, but alkyl and halo groups in the 1 position decrease the rates because of steric hindrance.
4. α Substitution. Compounds of the formula 2X, where Z = RO, RS, or R2N undergo SN1 reactions very rapidly,308 because of the increased resonance in the carbocation. These groups have an unshared pair on an atom directly attached to the positive carbon, which stabilizes the carbocation (Sec. 5.A.ii). The field effects of these groups would be expected to decrease SN1 rates (see Sec. 10.G.i, category 6), so the resonance effect is far more important.
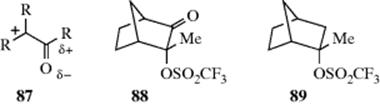
When Z in 2X is RCO,309 HCO, ROCO, NH2CO, NC, or F3C,310 SN1 rates are decreased compared to CH3X, owing to the electron-withdrawing field effects of these groups. Furthermore, carbocations311 with a CO or CN group are greatly destabilized because of the partial positive charge on the adjacent carbon (87). The SN1 reactions have been carried out on such compounds,312 but the rates are very low. For example, from a comparison of the solvolysis rates of 88 and 89, a rate-retarding effect of 1076 was estimated for the C=O group.313 However, when a different kind of comparison is made: RCOCR′2X versus HCR′2X (where X = a leaving group), the RCO had only a small or negligible rate-retarding effect, indicating that resonance314 may be offsetting the inductive destabilization for this group.315 For a CN group also, the rate-retarding effect is reduced by this kind of resonance.316A carbocation with a COR group has been isolated.317 When SN2 reactions are carried out on these substrates, rates are greatly increased for certain nucleophiles (e.g., halide or halide-like ions), but decreased or essentially unaffected by others.318 For example, α-chloroacetophenone (PhCOCH2Cl) reacts with KI in acetone at 75 °C ~ 32,000 times faster than 1-chlorobutane,319 but α-bromoacetophenone reacts with the nucleophile triethylamine 0.14 times as fast as iodomethane.318 The reasons for this varying behavior are not clear, but those nucleophiles that form a “tight” transition state (one in which bond making and breaking have proceeded to about the same extent) are more likely to accelerate the reaction.320
When Z is SOR or SO2R (e.g., α-halo sulfoxides and sulfones), nucleophilic substitution is retarded.321 The SN1 mechanism is slowed by the electron- withdrawing effect of the SOR or SO2R group,322 and the SN2 mechanism presumably by the steric effect.
5. β Substitution. For compounds of the type ZCH2CH2X, where Z is any of the groups listed in Section 10.F, as well as halogen323 or phenyl, SN1 rates are lower than for unsubstituted systems, because the resonance effects mentioned in item 4 are absent, but the field effects are still there, although smaller. These groups in the β position do not have much effect on SN2 rates unless they behave as neighboring groups and enhance the rate through anchimeric assistance,324 or unless their size causes the rates to decrease for steric reasons.325 It has been shown that silicon exerts a β-effect, and that tin exerts a γ-effect.326 Silcon also exerts a exerts γ-effect.327
6. The Effect of Electron-Donating and Electron-Withdrawing Groups. If substitution rates for a series of compounds p-ZC6H4CH2X are measured, it is possible to study the electronic effects of groups Z on the reaction. Steric effects of Z are minimized or eliminated, because Z is so far from the reaction site. For SN1 reactions, electron-withdrawing Z decrease the rate and electron-donating Z increase it,328 because the latter decrease the energy of the transition state (and of the carbocation) by spreading the positive charge, for example,
![]()
while electron-withdrawing groups concentrate the charge. The Hammett σρ relationship329 (Sec. 9.C) fairly successfully correlates the rates of many of these reactions (with σ+ instead of σ). The ρ values are generally about –4, which is expected for a reaction where a positive charge is created in the transition state.For SN2 reactions, no such simple correlations are found.330 In this mechanism, bond breaking is about as important as bond making in the rate-determining step, and substituents have an effect on both processes, often in opposite directions. The unsubstituted benzyl chloride and bromide solvolyze by the SN2 mechanism.322For Z = alkyl, the Baker–Nathan order(Sec. 2.M) is usually observed both for SN1 and SN2 reactions.In para-substituted benzyl systems, steric effects have been removed, but resonance and field effects are still present. However, Holtz and Stock331 studied a system that removes not only steric effects, but also resonance effects. This is the 4-substituted bicyclo[2.2.2]octylmethyl tosylate system (90). In this system, steric effects are completely absent owing to the rigidity of the molecules, and only field effects operate. By this means, Holtz and Stock showed that electron-withdrawing groups increase the rate of SN2 reactions. This can be ascribed to stabilization of the transition state by withdrawal of some of the electron density.
![]()
For substrates that react by the tetrahedral mechanism, electron-withdrawing groups increase the rate and electron-donating groups decrease it.
7. Cyclic Substrates. Cyclopropyl substrates are extremely resistant to nucleophilic attack.332 For example, cyclopropyl tosylate solvolyzes ~106 times more slowly than cyclobutyl tosylate in acetic acid at 60 °C.333 When such attack does take place, the result is generally not normal substitution (though exceptions are known,334 especially when a stabilizing group, e.g., aryl or alkoxy, is present), but ring opening:326
![]()
There is much evidence that the ring opening is usually concerted with the departure of the leaving group335 (as in the similar case of cyclobutyl substrates, Sec. 10.C.i, category 4.b.iv), from which we can conclude that if the 2,3-bond of the cyclopropane ring did not assist, the rates would be lower still. Strain plays a role in the ring-opening process.336 It has been estimated337 that without this assistance the rates of these already slow reactions would be further reduced by a factor of perhaps 1012. For a discussion of the stereochemistry of the ring opening, see Reaction 18-27, Section B. For larger rings, we have seen (Sec. 9.A) that, because of I strain, cyclohexyl substrates solvolyze slower than analogous compounds in which the leaving group is attached to a ring of 5 or of from 7 to 11 members.
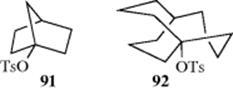
8. Bridgeheads.14 The SN2 mechanism is impossible at most bridgehead compounds (Sec. 10.A.i). Nucleophilic attack in [1.1.1]propellane has been reported, however.338 In general, a relatively large ring is required for an SN1 reaction to take place (Sec. 10.A.ii).339 The SN1 reactions have been claimed to occur for 1-iodobicyclo[1.1.1]pentane via the bicyclo[1.1.1]pentyl cation,340 but this has been disputed and the bicyclo[1.1.0]butylcarbinyl cation was calculated to be the real intermediate.341 Solvolytic reactivity at bridgehead positions spans a wide range; for example, from k = 4 × 10-17 s-1 for 91 (very slow) to 3 × 106 s-1 for the [3.3.3] compound 92 (very fast);342 a range of 22 orders of magnitude. Molecular mechanics calculations show that SN1 bridgehead reactivity is determined by strain changes between the substrate and the carbocation intermediate.343
9. Deuterium Substitution. Both α and β secondary isotope effects affect the rate in various ways (Sec. 6.J.vii). The measurement of a secondary isotope effect provides a means of distinguishing between SN1 and SN2 mechanisms, since for SN2 reactions the values range from 0.95 to 1.06 per α D, while for SN1 reactions the values are higher.344 This method is especially good because it provides the minimum of perturbation of the system under study; changing from α H to α D hardly affects the reaction, while other probes (e.g., changing a substituent or the polarity of the solvent) may have a much more complex effect.
Table 10.3 is an approximate listing of groups in order of SN1 and SN2 reactivity. Table 10.4 shows the main reactions that proceed by the SN2 mechanism (if R = primary or, often, secondary alkyl).
Table 10.3 List of Groups in Approximately Descending Order of Reactivity Toward SN1 and SN2 Reactionsa
SN1 Reactivity
SN2 Reactivity
Ar3CX
Ar3CX
Ar2CHX
Ar2CHX
ROCH2X, RSCH2X, R2NCH2X
ArCH2X
R3CX
ZCH2X
ArCH2X
–C=C–CH2X
–C=C–CH2X
RCH2X ~ RCHDX ~ RCHDCH2X
R2CHX
R2CHX
RCH2X ~ R3CCH2X
R3CX
RCHDX
ZCH2CH2X
RCHDCH2X
R3CCH2X
–C=C–C–X
–C=C–C–X
ZCH2X
ZCH2CH2X
ArX
ArX
Bridgehead-X
[2.2.1] Bridgehead-X
a. Here Z is RCO, HCO, ROCO, NH2CO, NC, or a similar group.
Table 10.4 The More Important Synthetic Reactions of Chapter 10 That Take Place by an SN2 Mechanism.a,b
Reaction Number
Reactions
10-1
![]()
10-8
![]()
10-9
10-10
![]()
10-12
![]()
10-14
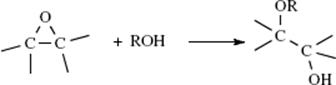
10-15
![]()
10-17
![]()
10-21
![]()
10-25
![]()
10-26
![]()
10-27
![]()
10-30
![]()
10-31
![]()
10-31
![]()
10-35
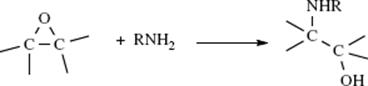
10-41
![]()
10-42
![]()
10-43
![]()
10-44
![]()
10-46
![]()
10-47
![]()
10-48
![]()
10-49
![]()
10-50
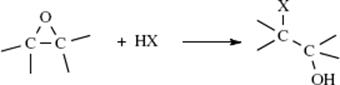
10-51
![]()
10-57
![]()
10-65
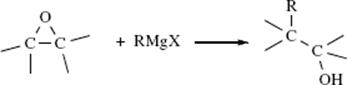
10-67
![]()
10-68
![]()
10-70
![]()
10-71
![]()
10-74
![]()
10-75
![]()
a. Here R = primary, often secondary, alkyl.
b. Catalysts are not shoure. This is a schematic list only. Some of these reactions may also take place by other mechanisms and the scope may vary greatly. See the discussion of each reaction for details.
10.G.ii The Effect of the Attacking Nucleophile345
Any species that has an unshared pair (i.e., any Lewis base) can, in principle, be a nucleophile, whether it is neutral or has a negative charge. The rates of SN1 reactions are independent of the identity of the nucleophile, since it does not appear in the rate-determining step.346 This may be illustrated by the effect of changing the nucleophile from H2O to −OH for a primary and a tertiary substrate. For methyl bromide, which reacts by an SN2 mechanism, the rate is multiplied > 5000 by the change to the more powerful nucleophile −OH, but for tert-butylbromide, which reacts by an SN1 mechanism, the rate is unaffected.347 A change in nucleophile can, however, change the product of an SN1 reaction. Thus solvolysis of benzyl tosylate in methanol gives benzyl methyl ether (the nucleophile is the solvent methanol). If the more powerful nucleophile Br− is added, the rate is unchanged, but the product is now benzyl bromide.
Note that the so-called cation affinity is used to measure the ability of a cation to interact with an electron-donating species. While this is not formally used to describe SN1 reactions, it is important for catalytic activity due to different ligands.348
For SN2 reactions in solution, there are four main principles that govern the effect of the nucleophile on the rate. The nucleophilicity order is not invariant, but depends on substrate, solvent, leaving group, and so on.
1. A nucleophile with a negative charge is always a more powerful nucleophile than its conjugate acid (assuming the latter is also a nucleophile). Thus −OH is more powerful than H2O, −NH2 is more powerful than NH3, and so on
2. In comparing nucleophiles whose attacking atom is in the same row of the periodic table, nucleophilicity is approximately the same as the order of basicity,349 although basicity is thermodynamically controlled and nucleophilicity is kinetically controlled. So an approximate order of nucleophilicity is −NH2 > RO− > −OH > R2NH > ArO− > NH3 > pyridine > F− > H2O > ClO4−, and another is R3C− > R2N− > RO− > F− (see Table 8.1). This type of correlation works best when the structures of the nucleophiles being compared are similar, as with a set of substituted phenoxides. Within such a series, linear relationships can often be established between nucleophilic rates and pK values.350
3. Going down the periodic table, nucleophilicity increases, although basicity decreases. Thus the usual order of halide nucleophilicity is I− > Br− > Cl− > F− (as seen below, this order is solvent dependent). Similarly, any sulfur nucleophile is more powerful than its oxygen analogue The same is true for phosphorus versus nitrogen. The main reason for this distinction between basicity and nucleophilic power is that the smaller negatively charged nucleophiles are more solvated by the usual polar protic solvents; that is, because the negative charge of Cl− is more concentrated than the charge of I−, the former is more tightly surrounded by a shell of solvent molecules that constitute a barrier between it and the substrate. This finding is most important for protic polar solvents in which the solvent may be hydrogen bonded to small nucleophiles. Evidence for this is that many nucleophilic substitutions with small negatively charged nucleophiles are much more rapid in aprotic polar solvents than in protic ones.351 Also, in DMF, an aprotic solvent, the order of nucleophilicity was Cl− > Br− > I−.352 Another experiment was the use of Bu4N+ X− and LiX as nucleophiles in acetone, where X− was a halide ion. The halide ion in the former salt is much less associated than in LiX. The relative rates with LiX were Cl−, 1−; Br−, 5.7; I−, 6.2, which is in the normal order, while with Bu4N+ X−, where X− is much freer, the relative rates were Cl−, 68; Br−, 18; ––, 3.7.353 In a further experiment, halide ions were allowed to react with the molten salt (n-C5H11)4N+ X−at 180 °C in the absence of a solvent.354 Under these conditions, where the ions are unsolvated and unassociated, the relative rates were Cl−, 620; Br−, 7.7; I−, 1. In the gas phase (no solvent), an approximate order of nucleophilicity was found to be ![]() > F− ~ MeO− > MeS− >> Cl− > −CN > Br−,355 providing further evidence that solvation356 is responsible for the effect in solution.
> F− ~ MeO− > MeS− >> Cl− > −CN > Br−,355 providing further evidence that solvation356 is responsible for the effect in solution.
However, solvation is not the entire answer since, even for uncharged nucleophiles, nucleophilicity increases going down a column in the periodic table. These nucleophiles are not so greatly solvated and changes in solvent do not greatly affect their nucleophilicity.357 To explain these cases, the principle of hard and soft acids and bases (Sec. 8.E) may be used.358 The proton is a hard acid, but an alkyl substrate (which may be considered to act as a Lewis acid toward the nucleophile considered as a base) is a good deal softer. According to the principle given in Section 8.F, an alkyl group is expected to prefer softer nucleophiles than the proton. Thus the larger, more polarizable (softer) nucleophiles have a greater (relative) attraction toward an alkyl carbon than toward a proton.
4. The freer the nucleophile, the greater the rate.359 One instance of this has already been discussed.353 Another is that the rate of attack by (EtOOC)2CBu− Na+ in benzene was increased by the addition of substances (e.g., 1,2-dimethoxyethane, adipamide) that specifically solvated the Na+ and thus left the anion freer.360 In a nonpolar solvent (e.g., benzene), salts, [e.g., (EtOOC)2CBu− Na+], usually exist as ion-pair aggregations of large molecular weights.361 Similarly, it was shown that the half-life of the reaction between C6H5COCHEt− and ethyl bromide depended on the positive ion: K+, 4.5 × 10−3; Na+, 3.9 × 10−5; Li+, 3.1 × 10−7.362 Presumably, the potassium ion leaves the negative ion freest to attack most rapidly. Further evidence is that in the gas phase,363 where nucleophilic ions are completely free, without solvent or counterion, reactions take place orders of magnitude faster than the same reactions in solution.364 It has proven possible to measure the rates of reaction of ![]() with methyl bromide in the gas phase, with
with methyl bromide in the gas phase, with ![]() either unsolvated or solvated with one, two, or three molecules of water.365 The rates were, with the number of water molecules in parentheses: (0) 1.0 × 10−9; (1) 6.3 × 10−10; (2) 2 × 10−12; (3) 2 × 10−13 cm3 molecule−1 s−1, evidence that solvation of the nucleophile decreases the rate. The rate of this reaction in aqueous solution is 2.3 × 10−25 cm3 molecule−1 s−1. Similar results were found for other nucleophiles and other solvents.366 Also, solution studies have been made of the effect of solvation of the nucleophile by a specific number of water molecules. Indeed, hydrogen bonding lowers the intrinsic nucleophilicity.367 When the salt (n−C6H13)4N+F− reacted with n-octyl methanesulfonate, the relative rate fell from 822 for no water molecules to 96 for 1.5 water molecules to 1 for 6 water molecules.368
either unsolvated or solvated with one, two, or three molecules of water.365 The rates were, with the number of water molecules in parentheses: (0) 1.0 × 10−9; (1) 6.3 × 10−10; (2) 2 × 10−12; (3) 2 × 10−13 cm3 molecule−1 s−1, evidence that solvation of the nucleophile decreases the rate. The rate of this reaction in aqueous solution is 2.3 × 10−25 cm3 molecule−1 s−1. Similar results were found for other nucleophiles and other solvents.366 Also, solution studies have been made of the effect of solvation of the nucleophile by a specific number of water molecules. Indeed, hydrogen bonding lowers the intrinsic nucleophilicity.367 When the salt (n−C6H13)4N+F− reacted with n-octyl methanesulfonate, the relative rate fell from 822 for no water molecules to 96 for 1.5 water molecules to 1 for 6 water molecules.368
In Chapter 3, cryptands were seen to specifically solvate the alkali metal portion of salts like KF, KOAc, and so on. Synthetic advantage can be taken of this fact to allow anions to be freer, thus increasing the rates of nucleophilic substitutions and other reactions (see Sec. 10.G.v).
However, the four rules given above do not always hold. One reason is that steric influences often play a part. For example, the tert-butoxide ion Me3CO− is a stronger base than ![]() or
or ![]() , but a much poorer nucleophile because its large bulk hinders it from closely approaching a substrate.
, but a much poorer nucleophile because its large bulk hinders it from closely approaching a substrate.
The following overall nucleophilicity order for SN2 mechanisms (in protic solvents) was given by Edwards and Pearson369: RS− > ArS− > I− >CN− > ![]() > N3− > Br− > ArO− > Cl− > pyridine > AcO− > H2O. A quantitative relationship370 (the Swain–Scott equation, which can be derived from Marcus theory371) has been worked out similar to the linear free-energy equations considered in Chapter 9:372
> N3− > Br− > ArO− > Cl− > pyridine > AcO− > H2O. A quantitative relationship370 (the Swain–Scott equation, which can be derived from Marcus theory371) has been worked out similar to the linear free-energy equations considered in Chapter 9:372
![]()
where n is the nucleophilicity of a given group, s is the sensitivity of a substrate to nucleophilic attack, and k0 is the rate for H2O, which is taken as the standard and for which n is assigned a value of zero. The parameter s is defined as 1.0 for bromomethane. Table 10.5 contains values of n for some common nucleophiles.373 The order is similar to that of Edwards and Pearson.
Table 10.5 Nucleophiliclties of Some Common Reagentsa
Reprinted with permission Wells, P.R. Chem. Rev. 1963, 63, 171. Copyright © 1963 American Chemical Society.
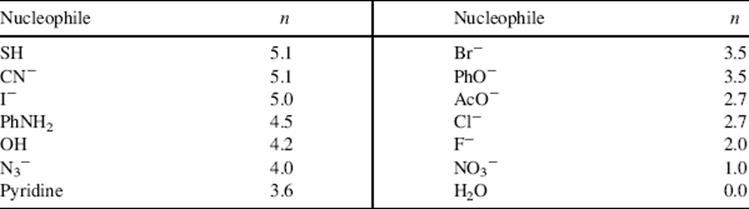
a See Ref. 373.
It is now evident that there is no absolute order of either nucleophilicity374 or leaving-group ability, even in the gas phase where solvation is not a factor because they have an effect on each other. When the nucleophile and leaving group are both hard or both soft, the reaction rates are relatively high, but when one is hard and the other soft, rates are reduced.363 Although this effect is smaller than the effects in paragraphs one and four above, it still prevents an absolute scale of either nucleophilicity or leaving-group ability.375 There has been controversy as to whether the selectivity of a reaction should increase with decreasing reactivity of a series of nucleophiles, or whether the opposite holds. There is evidence for both views.376
For substitution at a carbonyl carbon, the nucleophilicity order is not the same as it is at a saturated carbon, but follows the basicity order more closely. The reason is presumably that the carbonyl carbon has a partial positive charge. That is, a carbonyl carbon is a much harder acid than a saturated carbon. The following nucleophilicity order for these substrates has been determined:377 Me2C=NO− > EtO− > MeO− > ![]() >
> ![]() > N3− > F− > H2O > Br−~ I−. Soft bases are ineffective at a carbonyl carbon.378 In a reaction carried out in the gas phase with alkoxide nucleophiles OR− solvated by only one molecule of an alcohol R′OH, it was found that both RO− and R′O− attacked the formate substrate (HCO2R″) about equally, although in the unsolvated case, the more basic alkoxide is the better nucleophile.379 In this study, the product ion R2O− was also solvated by one molecule of ROH or R′OH.
> N3− > F− > H2O > Br−~ I−. Soft bases are ineffective at a carbonyl carbon.378 In a reaction carried out in the gas phase with alkoxide nucleophiles OR− solvated by only one molecule of an alcohol R′OH, it was found that both RO− and R′O− attacked the formate substrate (HCO2R″) about equally, although in the unsolvated case, the more basic alkoxide is the better nucleophile.379 In this study, the product ion R2O− was also solvated by one molecule of ROH or R′OH.
If an atom containing one or more unshared pairs is adjacent to the attacking atom on the nucleophile, the nucleophilicity is enhanced.380 Examples of such nucleophiles are HO2−, Me2C=NO−, NH2NH2, and so on. This is called the alpha effect (α-effect),381 and a broader definition is a positive deviation exhibited by an α-nucleophile from a Br![]() nsted-type nucleophilicity plot,382 where the reference (or normal) nucleophile is one that possesses the same basicity as the α-nucleophile, but does not deviate from the Br
nsted-type nucleophilicity plot,382 where the reference (or normal) nucleophile is one that possesses the same basicity as the α-nucleophile, but does not deviate from the Br![]() nsted-type plot. Several reviews of the α-effect have been published previously.72,383 Several possible explanations have been offered.384 One is that the ground state of the nucleophile is destabilized by repulsion between the adjacent pairs of electrons;385 another is that the transition state is stabilized by the extra pair of electrons;386 a third is that the adjacent electron pair reduces solvation of the nucleophile.387 Evidence supporting the third explanation is that there was no alpha effect in the reaction of HO2- with methyl formate in the gas phase,388 although HO2− shows a strong alpha effect in solution. The α-effect has been demonstrated to be remarkably dependent on the nature of the solvent.389 The α-effect is substantial for substitution at a carbonyl or other unsaturated carbon, at some inorganic atoms,390 and for reactions of a nucleophile with a carbocation,391 but is generally smaller or absent entirely for substitution at a saturated carbon.392
nsted-type plot. Several reviews of the α-effect have been published previously.72,383 Several possible explanations have been offered.384 One is that the ground state of the nucleophile is destabilized by repulsion between the adjacent pairs of electrons;385 another is that the transition state is stabilized by the extra pair of electrons;386 a third is that the adjacent electron pair reduces solvation of the nucleophile.387 Evidence supporting the third explanation is that there was no alpha effect in the reaction of HO2- with methyl formate in the gas phase,388 although HO2− shows a strong alpha effect in solution. The α-effect has been demonstrated to be remarkably dependent on the nature of the solvent.389 The α-effect is substantial for substitution at a carbonyl or other unsaturated carbon, at some inorganic atoms,390 and for reactions of a nucleophile with a carbocation,391 but is generally smaller or absent entirely for substitution at a saturated carbon.392
Attempts have been made to establish a general scale of nucleophilicity,393 and the nucleophilic reactivity of other moieties have been determined, including alcohols and alkoxides,394 carbanions,395 amines,396 pyridines,397pyrroles,398 indoles,399 imides and amides,400 amino acids and peptides,401 and sulfur ylids.402
10.G.iii. The Effect of the Leaving Group
The leaving group at the saturated carbon comes off more easily the more stable it is as a free entity. This is usually inverse to its basicity, and the best leaving groups are the weakest bases. Thus iodide is the best leaving group among the halides and fluoride is the poorest. Since XH is always a weaker base than X−, nucleophilic substitution is always easier at a substrate RXH+ than at RX. An example of this effect is that OH and OR are not leaving groups from ordinary alcohols and ethers, but can come off when the groups are protonated (i.e., converted to ROH2+ or RORH+).403 Reactions in which the leaving group does not come off until it has been protonated have been called SN1cA or SN2cA, depending on whether after protonation the reaction is an SN1 or SN2 process (these designations are often shortened to A1 and A2). The cA stands for conjugate acid, since the substitution takes place on the conjugate acid of the substrate. The IUPAC designations for these mechanisms are, respectively, Ah + DN + AN and Ah + ANDN; that is, the same designations as SN1 and SN2, with Ah to show the preliminary step. When another electrophile assumes the role of the proton, the symbol Ae is used instead. The ions ROH2+ and RORH+ can be observed as stable entities at low temperatures in superacid solutions.404 At higher temperatures, they cleave to give carbocations.
It is obvious that the best nucleophiles (e.g., NH2−, ![]() ) cannot take part in SN1cA or SN2cA processes, because they would be converted to their conjugate acids under the acidic conditions necessary to protonate the leaving groups.405 Because SN1 reactions do not require powerful nucleophiles, but do require good leaving groups, most of them take place under acidic conditions. In contrast, SN2 reactions, which do require powerful nucleophiles, which are generally strong bases, most often take place under basic or neutral conditions.
) cannot take part in SN1cA or SN2cA processes, because they would be converted to their conjugate acids under the acidic conditions necessary to protonate the leaving groups.405 Because SN1 reactions do not require powerful nucleophiles, but do require good leaving groups, most of them take place under acidic conditions. In contrast, SN2 reactions, which do require powerful nucleophiles, which are generally strong bases, most often take place under basic or neutral conditions.
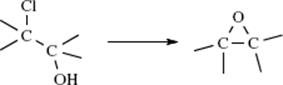
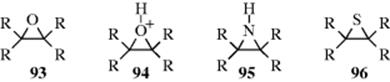
Another circumstance that increases leaving-group power is ring strain. Ordinary ethers do not cleave at all and protonated ethers only under strenuous conditions, but epoxides406 (93) are cleaved quite easily and protonated epoxides (94) even more easily. Aziridines (95)407 and episulfides (96) are also easily cleaved (see Sec. 10.G.viii).408
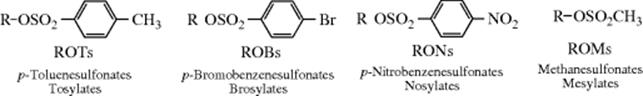
Although halides are common leaving groups in nucleophilic substitution for synthetic purposes, it is often more convenient to use alcohols. Since OH does not leave from ordinary alcohols, it must be converted to a group that does leave. One way is protonation, mentioned above. Another is conversion to a reactive ester, most commonly a sulfonic ester. The sulfonic ester groups tosylate, brosylate, nosylate, and mesylate are better leaving groups than halides and are frequently used.409 Other leaving groups are still better, and compounds containing these groups make powerful alkylating agents. Among them are oxonium ions (ROR2+),410 and the fluorinated compounds triflates411 and nonaflates.411Tresylates are ~ 400 times less reactive than triflates, but still ~ 100 times more reactive than tosylates.412 Halonium ions (RClR+, RBrR+, RIR+), which can be prepared in superacid solutions (Sec. 5.A.ii) and isolated as solid SbF6− salts, are also extremely reactive in nucleophilic substitution.413 Of the above types of compound, the most important in organic synthesis are tosylates, mesylates, oxonium ions, and triflates. The others have been used mostly for mechanistic purposes.
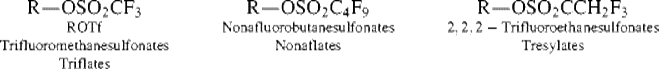
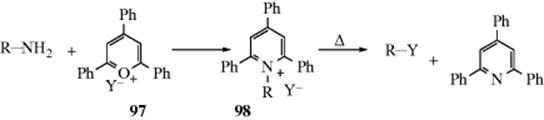
The leaving group ability of NH2, NHR, and NR2 are extremely poor,414 but the leaving-group ability of NH2 can be greatly improved by converting a primary amine (RNH2) to the ditosylate (RNTs2). The NTs2 group has been successfully replaced by a number of nucleophiles.415 Another way of converting NH2 into a good leaving group has been extensively developed by Katritzky et al.416 In this method, the amine is converted to a pyridinium compound (98) by treatment with a pyrylium salt (frequently a 2,4,6-triphenylpyrylium salt, 97).417 When the salt is heated, the counterion acts as a nucleophile. In some cases, a non-nucleophilic ion (e.g., BF4−), is used as the counterion for the conversion 97 → 98, and then Y− is added to 98. Among the nucleophiles that have been used successfully in this reaction are I−, Br−, Cl−, F−, ![]() , N3−, NHR2, and H−. Ordinary NR2 groups are good leaving groups when the substrate is a Mannich base (these are compounds of the form RCOCH2CH2NR2; see Reaction 16-19).418 The elimination–addition mechanism applies in this case.
, N3−, NHR2, and H−. Ordinary NR2 groups are good leaving groups when the substrate is a Mannich base (these are compounds of the form RCOCH2CH2NR2; see Reaction 16-19).418 The elimination–addition mechanism applies in this case.
Probably the best leaving group is N2 from the species RN2+, which can be generated in several ways,419 of which the two most important are the treatment of primary amines with nitrous acid (see Reaction 13-19)
![]()
and the protonation of diazo compounds420
![]()
No matter how produced, RN2+ are usually too unstable to be isolable,421 reacting presumably by the SN1 or SN2 mechanism.422 The simplest aliphatic diazonium ion (CH2N2+) has been prepared at −120 °C in superacid solution, where it lived long enough for an NMR spectrum to be taken.423 Actually, the exact mechanisms are in doubt because the rate laws, stereochemistry, and products have proved difficult to interpret.424 If there are free carbocations they should give the same ratio of substitution to elimination to rearrangements, and so on, as carbocations generated in other SN1 reactions, but they often do not. “Hot” carbocations (unsolvated and/or chemically activated) that can hold their configuration have been postulated,425 as have ion pairs, in which ![]() (or
(or ![]() , etc., depending on how the diazonium ion is generated) is the counterion.426 One class of aliphatic diazonium salts of which several members have been isolated as stable salts are the cyclopropeniumyldiazonium salts:427
, etc., depending on how the diazonium ion is generated) is the counterion.426 One class of aliphatic diazonium salts of which several members have been isolated as stable salts are the cyclopropeniumyldiazonium salts:427
Diazonium ions generated from ordinary aliphatic primary amines are usually useless for preparative purposes, since they lead to a mixture of products giving not only substitution by any nucleophile present, but also elimination and rearrangements if the substrate permits. For example, diazotization of n-butylamine gave 25% 1-butanol, 5.2% 1-chlorobutane, 13.2% 2-butanol, 36.5% butenes (consisting of 71% 1-butene, 20% trans-2-butene, and 9% cis-2-butene), and traces of butyl nitrites.428
In the SN1cA and SN2cA mechanisms (see above) there is a preliminary step, the addition of a proton, before the normal SN1 or SN2 process occurs. There are also reactions in which the substrate loses a proton in a preliminary step. In these reactions, there is a carbene intermediate.
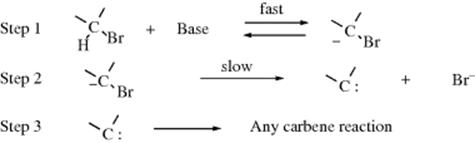
Once formed by this process, the carbene may undergo any of the normal carbene reactions (see Sec. 5.D.ii). When the net result is substitution, this mechanism has been called the SN1cB (for conjugate base) mechanism.429Although the slow step is an SN1 step, the reaction is second order; first order in substrate and first order in base.
Table 10.6 lists some leaving groups in approximate order of ability to leave.430–432430431432 The order of leaving-group ability is about the same for SN1 and SN2 reactions.
Table 10.6 Leaving Groups Listed in Approximate Order of Decreasing Ability to Leave.a
Common leaving groups
Substrate RX
At Saturated Carbon
At Carbonyl Carbon
RN2+
x
ROR'2+
ROSO2C4F9
ROSO2CF3
x
ROSO2F
ROTs, etc.b
x
RI
x
RBr
x
ROH2+
x (conjugate acid of alcohol)
RCl
x
x (acyl halides)
RORH+
x (conjugate acid of ether)
RONO2, etc.b
RSR'2+c
RNR'2+
x
RF
ROCOR′d
x
x (anhydrides)
RNH3+
ROAre
x (aryl esters)
ROH
x (carboxylic acids)
ROR
x (alkyl esters)
RH
RNH2
x (amides)
RAr
RR
a. Groups that are common leaving groups at saturated and carbonyl carbons are indicated;
b. The ROTs, and so on, includes esters of sulfuric and sulfonic acids in general, for example, ROSO2OH, ROSO2OR, ROSO2R, and so on, RONO2, and so on, includes inorganic ester leaving groups, such as ROPO(OH)2 and ROB(OH)2.
c. See Ref. 430.
d. See Ref. 431.
e. See Ref. 432.
10.G.iv. The Effect of the Reaction Medium433
The effect of solvent polarity434 on the rate of SN1 reactions depends on whether the substrate is neutral or positively charged.435 For neutral substrates, which constitute the majority of cases, the more polar the solvent, the faster the reaction, since there is a greater charge in the transition state than in the starting compound (Table 10.7436436) and the energy of an ionic transition state is reduced by polar solvents. However, when the substrate is positively charged, the charge is more spread out in the transition state than in the starting ion, and a greater solvent polarity slows the reaction. Even for solvents with about the same polarity, there is a difference between protic and aprotic solvents.437The SN1 reactions of un-ionized substrates are more rapid in protic solvents, which can form hydrogen bonds with the leaving group. Examples of protic solvents are water,438 alcohols, and carboxylic acids, while some polar aprotic solvents are DMF, DMSO,439 acetonitrile, acetone, sulfur dioxide, and HMPA [(Me2N)3PO].440 An algorithm has been developed to accurately calculate dielectric screening effects in solvents.441 The SN2 reactions have been done in ionic liquids (see Sec. 9.D.iii)442 and in supercritical carbon dioxide (see Sec. 9.D.ii).443
Table 10.7 Transition States for SN1 Reactions of Charged and Uncharged Substrates, and for SN2 Reactions of the Four Charge Typesa
Reactants and Transition States
Charge in the Transition State Relative to Starting Materials
How an Increase in Solvent Polarity Affects the Rate
Type I RX + Y− → Yδ−⋅R⋅Xδ−
Dispersed
Small decrease
Type II RX + Y− → Yδ+⋅R⋅Xδ−
Increased
Large increase
SN2
Type III RX + Y− → Yδ−⋅R⋅Xδ+
Decreased
Large decrease
Type IV RX + Y− → Yδ−⋅R⋅Xδ+
Dispersed
Small decrease
RX → Rδ+⋅Xδ−
Increased
Large increase
SN1
RX- → Rδ−⋅Xδ−
Dispersed
Small decrease
Adapted material from Structure and Mechanism in Organic Chemistry, 2d ed., Cornell University Press, Ithaca, NY, 1969, pp. 457–463., edited by Ingold, C.K. Copyright © 1969 by Cornell University. Used by permission of the publisher, Cornell University Press.
a. See Ref. 436.
For SN2 reactions, the effect of the solvent444 depends on which of the four charge types the reaction belongs to (preceding Sec. 10.A). In types I and IV, an initial charge is dispersed in the transition state, so the reaction is hindered by polar solvents. In type III, initial charges are decreased in the transition state, so that the reaction is even more hindered by polar solvents. Only type II, where the reactants are uncharged but the transition state has built up a charge, is aided by polar solvents. These effects are summarized in Table 10.7.436 Westaway445 proposed a “solvation rule” for SN2 reactions, which states that changing the solvent will not change the structure of the transition state for type I reactions, but will change it for type II reactions. The difference between protic and aprotic solvents must be considered for SN2 reactions as well.446 For reactions of types I and III the transition state is more solvated in polar aprotic solvents than in protic ones,447 while (as seen in Sec. 10.G.ii) the original charged nucleophile is less solvated in aprotic solvents448 (the second factor is generally much greater than the first449). So the change from, say, methanol to DMSO should greatly increase the rate. As an example, the relative rates at 25 °C for the reaction between MeI and Cl- were in MeOH, 1;351 in HCONH2 (still protic although a weaker acid), 12.5; in HCONHMe, 45.3; and HCONMe2, 1.2 × 106. The change in rate in going from a protic to an aprotic solvent is also related to the size of the attacking anion. Small ions are solvated best in protic solvents, since hydrogen bonding is most important for them, while large anions are solvated best in aprotic solvents (protic solvents have highly developed structures held together by hydrogen bonds; aprotic solvents have much looser structures, and it is easier for a large anion to be fitted in). So the rate of attack by small anions is most greatly increased by the change from a protic to an aprotic solvent. This may have preparative significance. The review articles in Ref. 431 have lists of several dozen reactions of charge types I and III in which yields are improved and reaction times reduced in polar aprotic solvents. Reaction types II and IV are much less susceptible to the difference between protic and aprotic solvents.
Since for most reactions SN1 rates go up and SN2 rates go down in solvents of increasing polarity, it is quite possible for the same reaction to go by the SN1 mechanism in one solvent and the SN2 in another. Table 10.8 is a list of solvents in order of ionizing power;450 a solvent high on the list is a good solvent for SN1 reactions. Trifluoroacetic acid, which was not studied by Smith et al.,450 has greater ionizing power451 than any solvent listed in Table 10.8. Because it also has very low nucleophilicity, it is an excellent solvent for SN1 solvolyses. Other good solvents for this purpose are 1,1,1-trifluoroethanol (CF3CH2OH), and 1,1,1,3,3,3-hexafluoro-2-propanol [(F3C)2CHOH].452
Table 10.8 Relative Rates of Ionization of p-Methoxyneophyl Toluenesulfonate in Various Solventsa
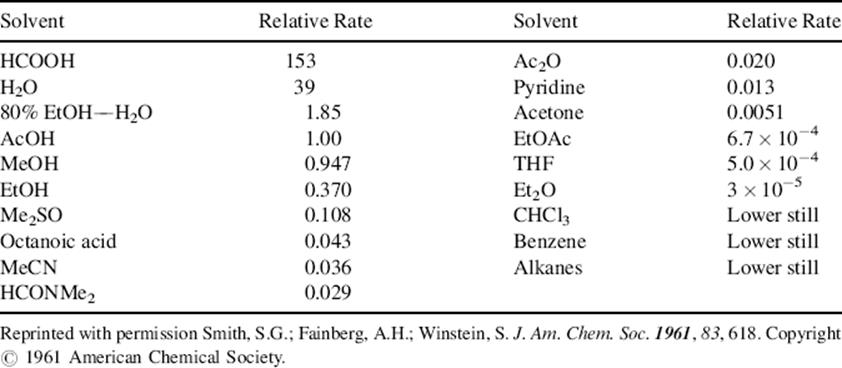
aSee Ref. 450.
Previously, the influence of the polarity of the solvent on the rates of SN1 and SN2 reactions was discussed. The ionic strength of the medium has similar effects. In general, the addition of an external salt affects the rates of SN1 and SN2 reactions in the same way as an increase in solvent polarity, although this is not quantitative; different salts have different effects.453 However, there are exceptions: Although the rates of SN1 reactions are usually increased by the addition of salts (this is called the salt effect), addition of the leaving-group ion often decreases the rate (the common-ion effect, Sec. 10.A.ii). There is also the special salt effect of LiClO4, mentioned on Section 10.A.iii, category 2. In addition to these effects, SN1 rates are also greatly accelerated when there are ions present that specifically help in pulling off the leaving group.454 Especially important are Ag+, Hg2+, and Hg22+, but H+ helps to pull off F (hydrogen bonding).455 Even primary halides have been reported to undergo SN1 reactions when assisted by metal ions.456 This does not mean, however, that reactions in the presence of metallic ions invariably proceed by the SN1 mechanism. It has been shown that alkyl halides can react with AgNO2 and AgNO3 by the SN1 or SN2 mechanism, depending on the reaction conditions.457
The effect of solvent has been treated quantitatively (for SN1 mechanisms, in which the solvent pulls off the leaving group) by a linear free energy relationship458
![]()
where m is characteristic of the substrate (defined as 1.00 for t-BuCl) and is usually near unity, Y is characteristic of the solvent and measures its “ionizing power”, and k° is the rate in a standard solvent, 80% aq ethanol at 25 °C. This is known as the Grunwald–Winstein equation, and its utility is at best limited. The Y values can of course be measured for solvent mixtures as well. This is one of the principal advantages of the treatment, since it is not easy otherwise to assign a polarity arbitrarily to a given mixture of solvents.459 The treatment is most satisfactory for different proportions of a given solvent pair. For wider comparisons, the treatment is not so good quantitatively, although the Y values do give a reasonably good idea of solvolyzing power.460 Table 10.9 contains a list of some Y values.461
Table 10.9 The Y, YOTs, Z, and ET (30) Values for Some Solventsa
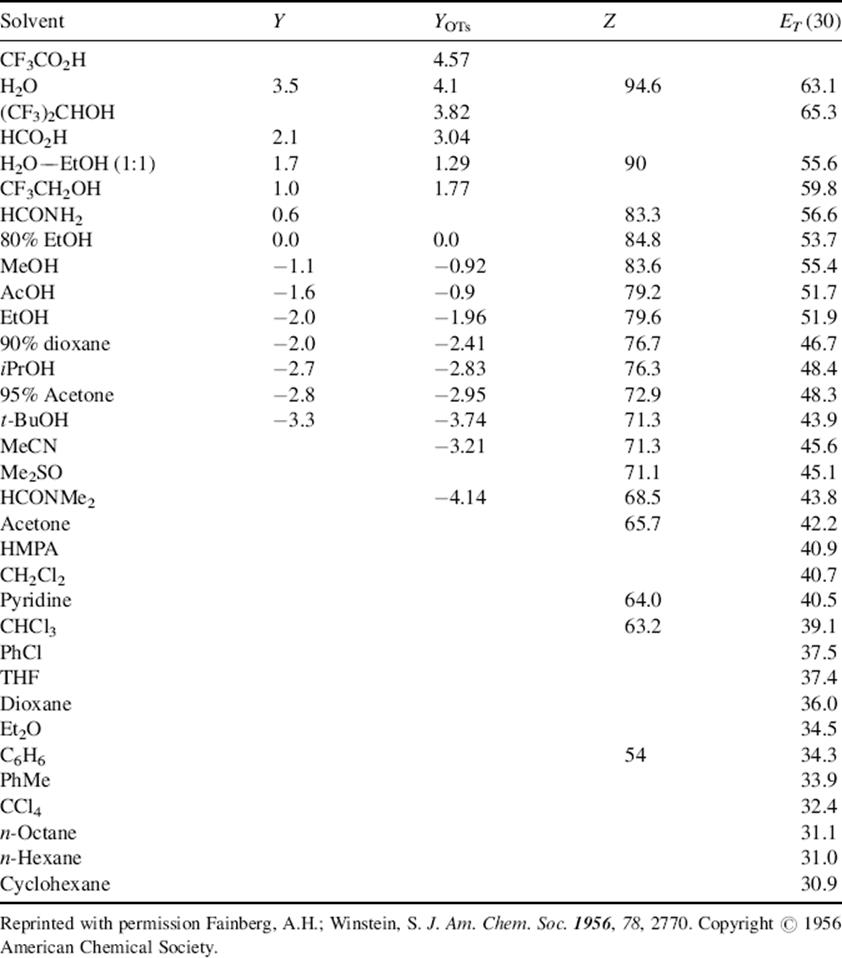
aSee Ref. 461.
Ideally, Y should measure only the ionizing power of the solvent, and should not reflect any backside attack by a solvent molecule in helping the nucleofuge to leave (nucleophilic assistance; ks, Sec. 10.C.i, category 3). Actually, there is evidence that many solvents do lend some nucleophilic assistance,462 even with tertiary substrates.463 It was proposed that a better measure of solvent “ionizing power” would be a relationship based on 2-adamantyl substrates, rather than t-BuCl, since the structure of this system completely prevents backside nucleophilic assistance (Sec. 10.G.i). Such a scale, called YOTs, was developed, with m defined as 1.00 for 2-adamantyl tosylate.464 Some values of YOTs are given in Table 10.9. These values, which are actually based on both 1- and 2-adamantyl tosylates (both are equally impervious to nucleophilic assistance and show almost identical responses to solvent ionizing power465) are called YOTs because they apply only to tosylates. It has been found that solvent “ionizing power” depends on the leaving group, so separate scales466 have been set up for OTf,467 Cl,433 Br,468 I,469 and other nucleofuges,470 all based on the corresponding adamantyl compounds. A new Y scale has been established based on benzylic bromides.471 In part, this was done because benzylic tosylates did not give a linear correlation with the 2-adamantyl YOTs parameter.472 This is substrate dependent, since solvolysis of 2,2-dimethyl-1-phenyl-1-propanol tosylate showed no nucleophilic solvent participation.473
In order to include a wider range of solvents than those in which any of the Y values can be conveniently measured, other attempts have been made at correlating solvent polarities.474 Kosower et al.475 found that the position of the charge-transfer peak (see Sec. 3.C.i) in the UV spectrum of the complex (99) between iodide ion and 1-methyl- or 1-ethyl-4-carbomethoxypyridinium ion was dependent on the polarity of the solvent.475 From these peaks, which are very easy to measure, Kosower et al.475 calculated transition energies that he called Z values. These values are thus measures of solvent polarity analogous to Y values. Another scale is based on the position of electronic spectra peaks of the pyridinium-N-phenolbetaine (100) in various solvents.476 Solvent polarity values on this scale are called ET(30)477 values. The ET(30) values are related to Z values by the expression478
![]()
Table 10.9 shows that Z and ET(30) values are generally in the same order as Y values. Other scales, the π∗ scale,479 the π∗azo scale,480 and the Py scale,481 are also based on spectral data.482
Carbon dioxide can be liquefied under high pressure (supercritical CO2). Several reactions have been done using supercritical CO2 as the medium (Sec. 9.D.ii), but special apparatus is required. This medium offers many advantages,483 and some disadvantages, but is an interesting new area of research.
The effect of solvent on nucleophilicity has already been discussed (Sec. 10.G.ii).
10.G.v. Phase-Transfer Catalysis
A difficulty that occasionally arises when carrying out nucleophilic substitution reactions is that the reactants do not mix. For a reaction to take place, the reacting molecules must collide. In nucleophilic substitutions, the substrate is usually insoluble in water and other polar solvents, while the nucleophile is often an anion, which is soluble in water, but not in the substrate or other organic solvents. Consequently, when the two reactants are brought together, their concentrations in the same phase are too low for convenient reaction rates. One way to overcome this difficulty is to use a solvent that will dissolve both species. As seen in Section 10.G.iv, a dipolar aprotic solvent may serve this purpose. Another way, which is used very often, is phase-transfer catalysis.484
In this method, a catalyst is used to carry the nucleophile from the aqueous into the organic phase. As an example, simply heating and stirring a two-phase mixture of 1-chlorooctane for several days with aqs NaCN gives essentially no yield of 1-cyanooctane. But if a small amount of an appropriate quaternary ammonium salt is added, the product is quantitatively formed in ~ 2 h.485 There are two principal types of phase-transfer catalyst, although the action of the two types is somewhat different, the effects are the same. Both get the anion into the organic phase and allow it to be relatively free to react with the substrate.
1. Quaternary Ammonium or Phosphonium Salts. In the above-mentioned case of NaCN, the uncatalyzed reaction does not take place because the ![]() ions cannot cross the interface between the two phases, except in very low concentration. The reason is that the Na+ ions are solvated by the water, and this solvation energy would not be present in the organic phase. The CN- ions cannot cross without the Na+ ions because that would destroy the electrical neutrality of each phase. In contrast to Na+ ions, quaternary ammonium (R4N+)486 and phosphonium (R4P+) ions with sufficiently large R groups are poorly solvated in water and prefer organic solvents. If a small amount of such a salt is added, three equilibria are set up:
ions cannot cross the interface between the two phases, except in very low concentration. The reason is that the Na+ ions are solvated by the water, and this solvation energy would not be present in the organic phase. The CN- ions cannot cross without the Na+ ions because that would destroy the electrical neutrality of each phase. In contrast to Na+ ions, quaternary ammonium (R4N+)486 and phosphonium (R4P+) ions with sufficiently large R groups are poorly solvated in water and prefer organic solvents. If a small amount of such a salt is added, three equilibria are set up:
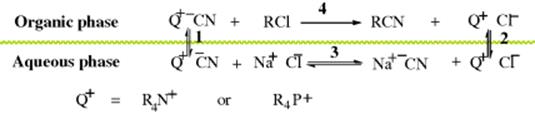
The Na+ ions remain in the aqueous phase; they cannot cross. The Q+ ions do cross the interface and carry an anion with them. At the beginning of the reaction, the chief anion present is ![]() . This gets carried into the organic phase (equilibrium 1) where it reacts with RCl to produce RCN and Cl−. The Cl− then gets carried into the aqueous phase (equilibrium 2). Equilibrium 3, taking place entirely in the aqueous phase, allows
. This gets carried into the organic phase (equilibrium 1) where it reacts with RCl to produce RCN and Cl−. The Cl− then gets carried into the aqueous phase (equilibrium 2). Equilibrium 3, taking place entirely in the aqueous phase, allows ![]() to be regenerated. All the equilibria are normally reached much faster than the actual conversion of RCl to RCN, so the latter is the rate-determining step.
to be regenerated. All the equilibria are normally reached much faster than the actual conversion of RCl to RCN, so the latter is the rate-determining step.
In some cases, the Q+ ions have such a low solubility in water that virtually all remain in the organic phase.487 In such cases, the exchange of ions (equilibrium 3) takes place across the interface. Still another mechanism (the interfacial mechanism) can operate where ![]() extracts a proton from an organic substrate.488 In this mechanism, the
extracts a proton from an organic substrate.488 In this mechanism, the ![]() ions remain in the aqueous phase and the substrate in the organic phase; the deprotonation takes place at the interface.489 Thermal stability of the quaternary ammonium salt is a problem, limiting the use of some catalysts. The trialkylacyl ammonium halide (101) is thermally stable, however, even at high reaction temperatures.490The use of molten quaternary ammonium salts as ionic reaction media for substitution reactions has also been reported.491
ions remain in the aqueous phase and the substrate in the organic phase; the deprotonation takes place at the interface.489 Thermal stability of the quaternary ammonium salt is a problem, limiting the use of some catalysts. The trialkylacyl ammonium halide (101) is thermally stable, however, even at high reaction temperatures.490The use of molten quaternary ammonium salts as ionic reaction media for substitution reactions has also been reported.491
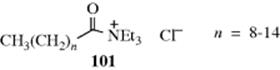
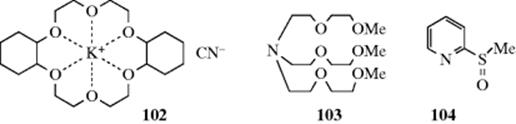
2. Crown Ethers and Other Cryptands.492 As seen in Section 3.C.ii, certain cryptands are able to surround certain cations. In effect, a salt like KCN is converted by dicyclohexano-18-crown-6 into a new salt (102) whose anion is the same, but whose cation is now a much larger species with the positive charge spread over a large volume, and hence much less concentrated. This larger cation is much less solubilized by water than K+ and much more attracted to organic solvents. Although KCN is generally insoluble in organic solvents, the cryptate salt is soluble in many of them. In these cases, we do not need an aqueous phase at all but simply add the salt to the organic phase. Suitable cryptands have been used to increase the rates of reactions where F−, Br−, I−, ![]() , and
, and ![]() are nucleophiles.493 Certain compounds that are not cryptands can act in a similar manner. One example is the podand tris(3,6-dioxaheptyl)amine (103), also called TDA-1.494 Another, not related to the crown ethers, is the pyridyl sulfoxide (104).495
are nucleophiles.493 Certain compounds that are not cryptands can act in a similar manner. One example is the podand tris(3,6-dioxaheptyl)amine (103), also called TDA-1.494 Another, not related to the crown ethers, is the pyridyl sulfoxide (104).495
Both of the above-mentioned catalyst types get the anions into the organic phase, but there is another factor as well. There is evidence that sodium and potassium salts of many anions, even if they could be dissolved in organic solvents, would undergo reactions very slowly (dipolar aprotic solvents are exceptions) because in these solvents the anions exist as ion pairs with Na+ or K+ and are not free to react with the substrate (Sec. 10.G.ii, category 4). Fortunately, ion pairing is usually much less with the quaternary ions and with the positive cryptate ions, so the anions in these cases are quite free to attack. Such anions are sometimes referred to as “naked” anions.
Not all quaternary salts and cryptands work equally well in all situations. Some experimentation is often required to find the optimum catalyst.
Although phase-transfer catalysis has been most often used for nucleophilic substitutions, it is not confined to these reactions. Any reaction that needs an insoluble anion dissolved in an organic solvent can be accelerated by an appropriate phase-transfer catalyst. Some examples will be seen in later chapters. In fact, in principle, the method is not even limited to anions, and a small amount of work has been done in transferring cations,496 radicals, and molecules.497 The reverse type of phase-transfer catalysis has also been reported: transport into the aqueous phase of a reactant that is soluble in organic solvents.498 Microwave activated phase-transfer catalysis has been reported.499
The catalysts mentioned above are soluble. Certain cross-linked polystyrene resins, as well as alumina500 and silica gel, have been used as insoluble phase-transfer catalysts. These, called triphase catalysts,501 have the advantage of simplified product work up and easy and quantitative catalyst recovery, since the catalyst can easily be separated from the product by filtration.
10.G.vi. Influencing Reactivity by External Means
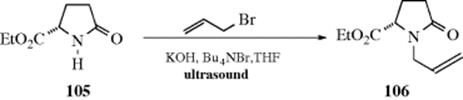
In many cases, reactions are slow. This is sometimes due to poor mixing or the aggregation state of one or more reactants. A powerful technique used to increase reaction rates is ultrasound (see Sec. 7.B). In this technique, the reaction mixture is subjected to high-energy sound waves, most often 20 KHz, but sometimes higher (a frequency of 20 KHz is about the upper limit of human hearing). When these waves are passed through a mixture, small bubbles form (cavitation). Collapse of these bubbles produces powerful shock waves that greatly increase the temperatures and pressures within these tiny regions, resulting in an increased reaction rate.502 In an instance where a metal, as a reactant or catalyst, is in contact with a liquid phase, a further effect is that the surface of the metal is cleaned and/or eroded by the ultrasound, allowing the liquid-phase molecules to come into closer contact with the metal atoms. Among the advantages of ultrasound is that it may increase yields, reduce side reactions, and permit the use of lower temperatures and/or pressures. The reaction of pyrrolidinone (105) with allyl bromide, under phase-transfer conditions, gave < 10% of the N-allyl product, (106). When the reaction was done under identical conditions, but with exposure to ultrasound (in an ultrasonic bath), the yield of 106 was 78%.503 It has been postulated that ultrasound has its best results with reactions that proceed, at least partially, through free radical intermediates.504
As noted in Chapter 7 (see Sec. 7.C), microwave irradiation is used extensively. Reaction times are greatly accelerated in many reactions, and reactions that took hours to be complete in refluxing solvents are done in minutes. Benzyl alcohol was converted to benzyl bromide, for example, using microwave irradiation (650 W) in only 9 min on a doped K-10 Montmorillonite clay.505 This technique is growing and very useful.
The rate of many reactions can be increased by application of high pressure.506 In solution, the rate of a reaction can be expressed in terms of the activation volume (ΔV)‡.507
![]()
The value of ΔV‡ is the difference in partial molal volume between the transition state and the initial state, but it can be approximated by the molar volume.507 Increasing pressure decreases the value of ΔV‡ and ΔV‡ is negative as the reaction rate is accelerated. This equation is not strictly obeyed > 10 kbar. If the transition state of a reaction involves bond formation, concentration of charge, or ionization, a negative volume of activation often results. Cleavage of a bond, dispersal of charge, neutralization of the transition state, and diffusion control lead to a positive volume of activation. Reactions for which rate enhancement is expected at high pressure include507:
1. Reactions in which the number of molecules decreases when starting materials are converted to products: cycloadditions [e.g., the Diels–Alder (Reaction 15-60)] and condensations, [e.g., the Knoevenagel condensation (16-38)].
2. Reactions that proceed via cyclic transition states: Claisen (Reaction 18-33) and Cope (Reaction 18-32) rearrangements.
3. Reactions that take place through dipolar transition states: Menshutkin reaction (Reaction 10-31), electrophilic aromatic substitution.
4. Reactions with steric hindrance.
Many high-pressure reactions are done neat, but if a solvent is used, the influence of pressure on that solvent is important. The melting point generally increases at elevated pressures, which influences the viscosity of the medium (viscosity of liquids increases approximately two times per kilobar increase in pressure). Controlling the rate of diffusion of reactants in the medium is also important.508 In most reactions, pressure is applied (5–20 kbar) at room temperature and then the temperature is increased until reaction takes place.
10.G.vii. Ambident (Bidentant) Nucleophiles: Regioselectivity
Some nucleophiles have a pair of electrons on each of two or more atoms, or canonical forms can be drawn in which two or more atoms bear an unshared pair. In these cases, the nucleophile may attack in two or more different ways to give different products. Such reagents are called ambident nucleophiles.509 In most cases, a nucleophile with two potentially attacking atoms can attack with either of them, depending on conditions, and mixtures are often obtained, although this is not always the case. For example, the nucleophile (NCO−) usually gives only isocyanates (RNCO) and not the isomeric cyanates (ROCN).510 When a reaction can potentially give rise to two or more structural isomers (e.g., ROCN or RNCO), but actually produces only one, the reaction is said to be regioselective511 (cf. the definitions of stereoselective, Sec. 4.N and enantioselective, Sec. 4.H., category 2). Some important ambident nucleophiles follow:
1. Ions of the Type –CO–−CR–CO–. These ions, which are derived by removal of a proton from malonic esters, β-keto esters, β-diketones, and so on, are resonance hybrids:
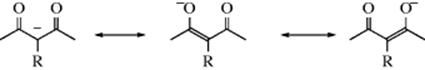
Attack is therefore possible at a saturated carbon via the carbon atoms (C-alkylation) or the oxygen atoms (O-alkylation):
With unsymmetrical ions, three products are possible, since either oxygen can attack. With a carbonyl substrate the ion can analogously undergo C- or O-acylation.
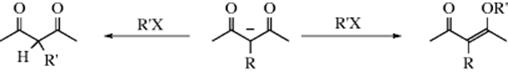
2. Compounds of the Type CH3CH–CH2–CO–. Can Give Up Two Protons, if treated with 2 molar equivalents of a strong enough base, to give dicarbanions:
![]()
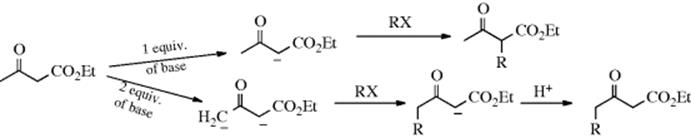
Such ions are ambident nucleophiles, since they have two possible attacking carbon atoms, aside from the possibility of attack by oxygen. In such cases, the attack is virtually always by the more basic carbon.512 Since the hydrogen of a carbon bonded to two carbonyl groups is more acidic than that of a carbon bonded to just one (see Chap 8), the CH group of 107 is less basic than the CH2 group, so the latter attacks the substrate. This gives rise to a useful general principle: Whenever the goal is to remove a proton at a given position for use as a nucleophile, but there is a stronger acidic group in the molecule, it may be possible to take off both protons; if it is, then attack is always by the desired position since it is the ion of the weaker acid. On the other hand, if the goal is to attack with the more acidic position, all that is necessary is to remove just one proton.513 For example, ethyl acetoacetate can be alkylated at either the methyl or the methylene group (Reaction 10-67):
3. The CN−Ion. This nucleophile can give nitriles (RCN, Reaction 10-75) or isocyanides (RN![]() C).
C).
4. The Nitrite Ion. This ion can give nitrite esters R–O–N=O (Reaction 10-22) or nitro compounds RNO2 (Reaction 10-76), which are not esters.
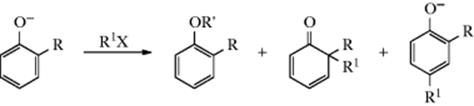
5. Phenoxide ions. These ions, (which are analogous to enolate anions) can undergo C- or O-alkylation:
6. Removal of a Proton from an Aliphatic Nitro Compound. This reaction gives a carbanion (R2C−–NO2) that can be alkylated at oxygen or carbon.514O-Alkylation gives a nitronic ester, and such compounds are generally unstable to heat and break down to give an oxime and an aldehyde or ketone.
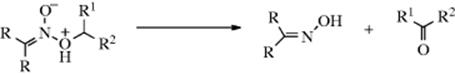
There are many other ambident nucleophiles.
It would be useful to have general rules as to which atom of an ambident nucleophile will attack a given substrate under a given set of conditions.515 Unfortunately, the situation is complicated by the large number of variables. It might be expected that the more electronegative atom would always attack, but this is often not the case. Where the products are determined by thermodynamic control (Sec. 6.F), the principal product is usually the one in which the atom of higher basicity has attacked (i.e., C > N > O > S).516 However, in most reactions, the products are kinetically controlled and matters are much less simple. Nevertheless, the following generalizations can be made, while recognizing that there are many exceptions and unexplained results. As in the discussion of nucleophilicity in general (Sec. 10.G.ii), there are two major factors: the polarizability (hard–soft character) of the nucleophile and solvation effects.
1. The principle of hard and soft acids and bases states that hard acids prefer hard bases and soft acids prefer soft bases (Sec. 8.E.i). In an SN1 mechanism, the nucleophile attacks a carbocation, which is a hard acid. In an SN2 mechanism, the nucleophile attacks the carbon atom of a molecule, which is a softer acid. The more electronegative atom of an ambident nucleophile is a harder base than the less electronegative atom. Therefore, as the character of a given reaction changes from SN1 to SN2 like, an ambident nucleophile becomes more likely to attack with its less electronegative atom.517 Thus, changing from SN1 to SN2 conditions should favor C attack by ![]() , N attack by NO2−, C attack by enolate or phenoxide ions, and so on. As an example, primary alkyl halides are attacked (in protic solvents) by the carbon atom of the anion of CH3COCH2CO2Et, while α-chloro ethers, which react by the SN1 mechanism, are attacked by the oxygen atom. However, this does not mean that attack is by the less electronegative atom in all SN2 reactions and by the more electronegative atom in all SN1 reactions. The position of attack also depends on the nature of the nucleophile, the solvent, the leaving group, and other conditions. The rule merely states that increasing the SN2 character of the transition state makes attack by the less electronegative atom more likely.
, N attack by NO2−, C attack by enolate or phenoxide ions, and so on. As an example, primary alkyl halides are attacked (in protic solvents) by the carbon atom of the anion of CH3COCH2CO2Et, while α-chloro ethers, which react by the SN1 mechanism, are attacked by the oxygen atom. However, this does not mean that attack is by the less electronegative atom in all SN2 reactions and by the more electronegative atom in all SN1 reactions. The position of attack also depends on the nature of the nucleophile, the solvent, the leaving group, and other conditions. The rule merely states that increasing the SN2 character of the transition state makes attack by the less electronegative atom more likely.
2. All negatively charged nucleophiles must of course have a positive counterion. If this ion is Ag+ (or some other ion that specifically helps in removing the leaving group, Sec. 10.G.iv), rather than the more usual Na+ or K+, then the transition state is more SN1 like. Therefore the use of Ag+ promotes attack at the more electronegative atom. For example, alkyl halides treated with NaCN generally give mostly RCN, but the use of AgCN increases the yield of isocyanides (RNC).518
3. In many cases, the solvent influences the position of attack. The freer the nucleophile, the more likely it is to attack with its more electronegative atom, but the more this atom is encumbered by either solvent molecules or positive counterions, the more likely it is to attack by the less electronegative atom. In protic solvents, the more electronegative atom is better solvated by hydrogen bonds than the less electronegative atom. In polar aprotic solvents, neither atom of the nucleophile is greatly solvated, but these solvents are very effective in solvating cations. Thus in a polar aprotic solvent the more electronegative end of the nucleophile is freer from entanglement by both the solvent and the cation, so that a change from a protic to a polar aprotic solvent often increases the extent of attack by the more electronegative atom. An example is attack by sodium β-naphthoxide on benzyl bromide, which resulted in 95% O-alkylation in DMSO and 85% C-alkylation in 2,2,2-trifluoroethanol.519 Changing the cation from Li+ to Na+ to K+ (in nonpolar solvents) also favors O- over C-alkylation520 for similar reasons (K+leaves the nucleophile much freer than Li+), as does the use of crown ethers, which are good at solvating cations (Sec. 3.C.ii).521 Alkylation of the enolate anion of cyclohexanone in the gas phase, where the nucleophile is completely free, showed only O-alkylation and no C-alkylation.522
4. In extreme cases, steric effects can govern the regioselectivity.523
10.G.viii. Ambident Substrates
Some substrates (e.g., 1,3-dichlorobutane) can be attacked at two or more positions, and these may be called ambident substrates. In the example given, there happen to be two leaving groups in the molecule. Apart from dichlorobutane, and in general, there are two kinds of substrates that are inherently ambident (unless symmetrical). One of these, the allylic type, has already been discussed (Sec. 10.E). The other is the epoxy (or the similar aziridine524 or episulfide) substrate.525 Selectivity for one or the other position is usually called regioselectivity.
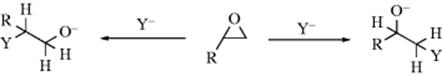
Substitution of the free epoxide, which generally occurs under basic or neutral conditions, usually involves an SN2 mechanism. Since primary substrates undergo SN2 attack more readily than secondary, unsymmetrical epoxides are attacked in neutral or basic solution at the less highly substituted carbon, and stereospecifically, with inversion at that carbon. Under acidic conditions, it is the protonated epoxide that undergoes the reaction. Under these conditions the mechanism can be either SN1 or SN2. In SN1 mechanisms, which favor tertiary carbons, attack may be expected be at the more highly substituted carbon, and this is indeed the case. However, even when protonated epoxides react by what is expected to be an SN2 mechanism, attack is usually at the more highly substituted position.526 This result probably indicates significant carbocation character at the carbon (ion pairing, for example). Thus, it is often possible to change the direction of ring opening by changing the conditions from basic to acidic or vice versa. In the ring opening of 2,3-epoxy alcohols, the presence of Ti(OiPr)4 increases both the rate and the regioselectivity, favoring attack at C-3 rather than C-2.527 When an epoxide ring is fused to a cyclohexane ring, SN2 ring opening invariably gives diaxial rather than diequatorial ring opening.528
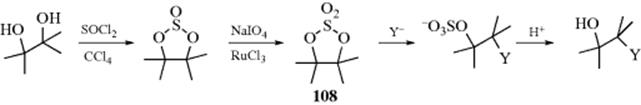
Cyclic sulfates (108), prepared from 1,2-diols, react in the same manner as epoxides, but usually more rapidly:529