March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 12. Aliphatic, Alkenyl, and Alkynyl Substitution, Electrophilic and Organometallic
Chapter 11 pointed out that the most important leaving groups in electrophilic substitution are those that can best exist with an outer shell that is deficient in a pair of electrons. For aromatic systems, the most common leaving group is the proton. The proton is also a leaving group in aliphatic systems, but the reactivity depends on the acidity. Protons in saturated alkanes are very unreactive, but electrophilic substitutions are often easily carried out at more acidic positions [e.g., α to a carbonyl group or at an alkynyl position (RC![]() CH)]. Since metallic ions are easily able to bear positive charges, organometallic compounds should be especially susceptible to electrophilic substitution, and this is indeed the case.1 Another important type of electrophilic substitution, known as anionic cleavage, involves the breaking of C–C bonds; in these reactions there are carbon leaving groups (Reactions 12-40–12-46). A number of electrophilic substitutions at a nitrogen atom are treated at the end of the chapter.
CH)]. Since metallic ions are easily able to bear positive charges, organometallic compounds should be especially susceptible to electrophilic substitution, and this is indeed the case.1 Another important type of electrophilic substitution, known as anionic cleavage, involves the breaking of C–C bonds; in these reactions there are carbon leaving groups (Reactions 12-40–12-46). A number of electrophilic substitutions at a nitrogen atom are treated at the end of the chapter.
Since a carbanion is generated when an atom or group is removed as a positive species from a carbon atom, the subject of carbanion structure and stability (Chapter 5) is inevitably related to the material in this chapter. So is the subject of very weak acids and very strong bases (Chapter 8), because the weakest acids are those in which the hydrogen is bonded to carbon.
12.A. Mechanisms
For aliphatic electrophilic substitution, at least four possible major mechanisms can be distinguished:2 SE1, SE2 (front), SE2 (back), and SEi. The SE1 is unimolecular; the other three are bimolecular. Note that the term “SEAr” has been proposed to represent electrophilic aromatic substitution, so that the term “SE2” refers exclusively to electrophilic substitutions where a steric course is possible.3 To describe the steric course of an aliphatic substitution reaction, the suffixes “ret” and “inv” were proposed, referring to retention and inversion of configuration, respectively.
12.A.i. Bimolecular Mechanisms: SE2 and SEi
The bimolecular mechanisms for electrophilic aliphatic substitution are analogous to the SN2 mechanism in that the new bond forms as the old one breaks. However, in the SN2 mechanism the incoming group brings with it a pair of electrons, and this orbital can overlap with the central carbon only to the extent that the leaving group takes away its electrons; otherwise the carbon would have more than eight electrons at once in its outer shell. Since electron clouds repel, this also means that the incoming group attacks backside, at a position 180 ° from the leaving group, resulting in inversion of configuration. When the nucleophilic species attacks (donates electrons to) an electrophile, it brings only a vacant orbital to the substrate. Predicting the direction of the attack is not as straightforward. Two main possibilities can be imagined: delivery of the electrophile to the front, which is SE2 (front), or delivery of the electrophile to the rear, which is SE2 (back). The possibilities can be pictured (charges not shown):
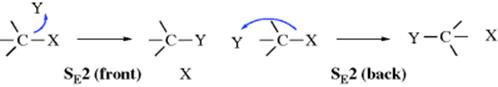
Both the SE2 (front) and SE2 (back) mechanisms are designated DEAE in the IUPAC system. With substrates in which these possibilities may be distinguished, the former mechanism should result in retention of configuration and the latter results in inversion. The reaction of allylsilanes with adamantyl chloride and TiCl4, for example, gives primarily the antiproduct via a SE2′ reaction.4 When the electrophile reacts from the front, there is a third possibility. A portion of the electrophile may assist in the removal of the leaving group, forming a bond with it at the same time that the new C–Y bond is formed:
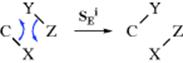
This mechanism, which is called the SEi mechanism5 (IUPAC designation: cyclo-DEAEDnAn), also results in retention of configuration.6 Plainly, where a second-order mechanism involves this kind of internal assistance, backside attack is impossible.
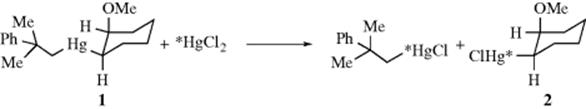
It is evident that these three mechanisms are not easy to distinguish. All three mechanisms give second-order kinetics, and two mechanisms result in retention of configuration.7 In fact, although much work has been done on this question, there are few cases in which one of these three can be unequivocally established to demonstrate that another is not actually taking place. Clearly, a study of the stereochemistry can distinguish between SE2 (back) on the one hand and SE2 (front) or SEi on the other. Many such investigations have been made. In the overwhelming majority of second-order electrophilic substitutions, the result has been retention of configuration or some other indication of frontside attack, indicating an SE2 (front) or SEi mechanism. For example, when cis-1 was treated with labeled mercuric chloride, the 2 produced was 100% cis. The bond between the mercury and the ring must have been broken (as well as the other Hg–C bond), since each of the products contained about one-half of the labeled mercury.8 Another indication of frontside attack is that second-order electrophilic substitutions proceed very easily at bridgeheadcarbons (see Sec. 10.A.i).9 Still another indication is the behavior of neopentyl as a substrate. The SN2 reactions at neopentyl are extremely slow (Sec. 10.G.i), because attack from the rear is blocked and the transition state for the reaction lies very high in energy. The fact that neopentyl systems undergo electrophilic substitution only slightly more slowly than ethyl10 is further evidence for frontside attack. One final elegant experiment may be noted.
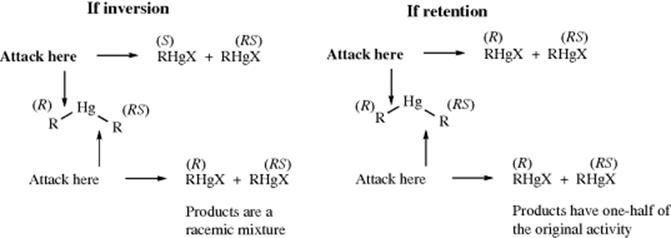
The compound di-sec-butylmercury was prepared with one sec-butyl group optically active and the other racemic.11 This compound was prepared by treatment of optically active sec-butylmercuric bromide with racemic sec-butylmagnesium bromide. The di-sec-butyl compound was then treated with mercuric bromide to give 2 molar equivalents of sec-butylmercuric bromide. The steric course of the reaction could then be predicted by the following analysis, assuming that the bonds between the mercury and each carbon have a 50% chance of breaking. The original activity referred to is the activity of the optically active sec-butylmercuric bromide used to make the dialkyl compound. The actual result was that, under several different sets of conditions, the product had one-half of the original activity, demonstrating retention of configuration.
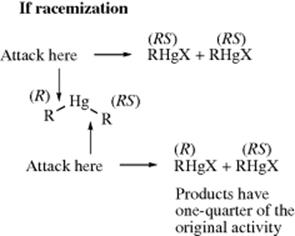
However, inversion of configuration has been found in certain cases, demonstrating that the SE2 (back) mechanism can take place. For example, the reaction of optically active sec-butyltrineopentyltin with bromine (Reaction 12-40) gives inverted sec-butyl bromide.12 A number of other organometallic compounds have also been shown to give inversion when treated with halogens,13 although others do not.14 So far, no inversion has been found with an organomercury substrate. It may be that still other examples of backside
![]()
attack exist15 but have escaped detection because of the difficulty in preparing compounds with a configurationally stable carbon–metal bond. Compounds that are chiral because of a stereogenic carbon at which a carbon–metal bond is located16 are often difficult to resolve and once resolved are often easily racemized. The resolution has been accomplished most often with organomercury compounds,17 and most stereochemical investigations have therefore been made with these substrates. Only a few optically active Grignard reagents (see Sec. 12-38) have been prepared18 (i.e., in which the only stereogenic center is the carbon bonded to the magnesium). Because of this, the steric course of electrophilic substitutions at the C–Mg bond has not often been determined. However, in one such case, the reaction of both the exo and endo isomers of the 2-norbornyl Grignard reagent with HgBr2 (to give 2-norbornylmercuric bromide) has been shown to proceed with retention of configuration.19 It is likely that inversion takes place only when steric hindrance prevents reaction on the frontside and when the electrophile does not carry a Z group (see above).
The SE2 (back) mechanism can be identified in certain cases (if inversion of configuration is found), but it is plain that stereochemical investigations cannot distinguish between the SE2 (front) and the SEi mechanisms and that, in the many cases where configurationally stable substrates cannot be prepared, such investigations are of no help at all in distinguishing among all three of the second-order mechanisms. Unfortunately, there are not many other methods that lead to unequivocal conclusions. One method that has been used in an attempt to distinguish between the SEi mechanism on the one hand and the SE2 pathways on the other, involves the study of salt effects on the rate. It may be recalled (Sec. 10.G.iv) that reactions in which neutral starting molecules acquire charges in the transition state are aided by an increasing concentration of added ions. Thus the SEi mechanism would be less influenced by salt effects than would either of the SE2 mechanisms. On this basis, Abraham and Johnson20 concluded that the reactions R4Sn + HgX2 → RHgX + R3SnX (X = Cl or I) take place by SE2 and not by SEi mechanisms. Similar investigations involve changes in solvent polarity21 (see also, Sec. 12.C.i). In the case of the reaction (where R = R′ = iPr and R = iPr, R′ = neopentyl), the use of polar solvents gave predominant inversion, while nonpolar solvents gave predominant retention.22
![]()
On the basis of evidence from reactivity studies, it has been suggested23 that a variation of the SEi mechanism is possible in which the group Z becomes attached to X before the latter becomes detached:
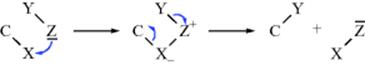
This process has been called the SEC22 or SE2 (co-ord)24 mechanism (IUPAC designation An + cyclo-DEAEDn).
It has been shown that in certain cases (e.g., Me4Sn + I2) the reactants in an SE2 reaction, when mixed, give rise to an immediate charge-transfer spectrum (Sec. 3.C.i), showing that an EDA complex has been formed.25 In these cases, it is likely that the EDA complex is an intermediate in the reaction.
12.A.ii. The SE1 Mechanism
The SE1 mechanism is analogous to the SN1. It involves two steps: A slow ionization and a fast combination.
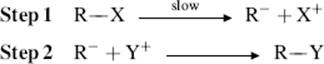
The IUPAC designation is DE + AE. First-order kinetics are predicted and many such examples have been found. Other evidence for the SE1 mechanism was obtained in a study of base-catalyzed tautomerization. In the reaction, the rate of deuterium exchange was the same as the rate of racemization26 and there was an isotope effect.27
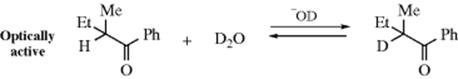
It is known that SN1 reactions do not proceed at strained bridgehead carbons (e.g., in [2.2.1]bicyclic systems, Sec. 10.A.ii) because planar carbocations cannot form at these carbons. However, carbanions not stabilized by resonance are probably not planar, and SE1 reactions readily occur with this type of substrate. Indeed, the question of carbanion structure is intimately tied into the problem of the stereochemistry of the SE1 reaction. If a carbanion is planar, racemization should occur. If it is pyramidal and can hold its structure, the result should be retention of configuration, or at least partial retention. On the other hand, even a pyramidal carbanion will give racemization if it cannot hold its structure (this means that there is pyramidal inversion as with amines, Sec. 4.C, category 3). Unfortunately, the only carbanions that can be studied easily are those stabilized by resonance, which makes them planar, as expected (Sec. 5.B.i). For simple alkyl carbanions, the main approach to deduce the structure has been to study the stereochemistry of SE1 reactions rather than the other way around. Racemization is almost always observed, but whether this is caused by planar carbanions or by oscillating pyramidal carbanions is not known. In either case, racemization occurs whenever a carbanion is completely free or is symmetrically solvated.
However, even planar carbanions need not give racemization. Cram found that retention and even inversion can occur in an alkoxide (see 3) cleavage Reaction (12-41):
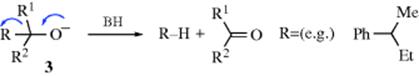
which is a first-order SE1 reaction involving resonance-stabilized planar carbanions (here designated R−).28 By changing the solvent, Cram was able to produce products ranging from 99% retention to 60% inversion and including complete racemization. These results are explained by a carbanion that is not completely free, but is solvated. In nondissociating, nonpolar solvents (e.g., benzene or dioxane), the alkoxide ion exists as an ion pair, solvated by the solvent BH:
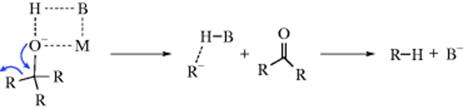
In the course of the cleavage, the proton of the solvent moves in to solvate the newly forming carbanion. This solvation is asymmetrical since the solvent molecule is already on the front side of the carbanion. When the carbanion actually bonds with the proton, the result is retention of the original configuration. In protic solvents (e.g., diethylene glycol), a good deal of inversion is found. In these solvents, the leaving group solvates the carbanion, so the solvent can solvate it only from the opposite side:
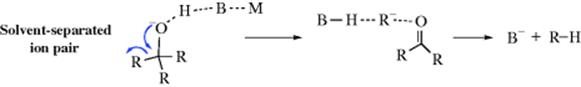
When C–H bond formation occurs, the result is inversion. Racemization occurs in polar aprotic solvents (e.g., DMSO). In these solvents, the carbanions are relatively long lived (because the solvent has no proton to donate) and is solvated symmetrically.
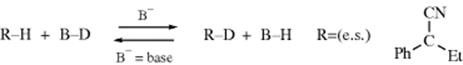
Similar behavior was found for carbanions generated by base-catalyzed hydrogen-exchange (Reaction 12-1).29 In this case, information was obtained from measurement of the ratio of ke (rate constant for isotopic exchange) to ka(rate constant for racemization). A keka ratio substantially >1 means retention of configuration, since many individual isotopic exchanges are not producing a change in configuration. A keka ratio of ~1 indicates racemization and a ratio of ![]() corresponds to inversion (see Sec. 10.A.i). All three types of steric behavior were found, depending on R, the base, and the solvent. As with the alkoxide cleavage reaction, retention was generally found in solvents of low dielectric constant, racemization in polar aprotic solvents, and inversion in protic solvents. However, in the proton-exchange reactions, a fourth type of behavior was encountered. In aprotic solvents, with aprotic bases like tertiary amines, the ke/ka ratio was found to be <0.5, indicating that racemization took place faster than isotopic exchange (this process is known as isoracemization). Under these conditions, the conjugate acid of the amine remains associated with the carbanion as an ion pair. Occasionally, the ion pair dissociates long enough for the carbanion to turn over and recapture the proton:
corresponds to inversion (see Sec. 10.A.i). All three types of steric behavior were found, depending on R, the base, and the solvent. As with the alkoxide cleavage reaction, retention was generally found in solvents of low dielectric constant, racemization in polar aprotic solvents, and inversion in protic solvents. However, in the proton-exchange reactions, a fourth type of behavior was encountered. In aprotic solvents, with aprotic bases like tertiary amines, the ke/ka ratio was found to be <0.5, indicating that racemization took place faster than isotopic exchange (this process is known as isoracemization). Under these conditions, the conjugate acid of the amine remains associated with the carbanion as an ion pair. Occasionally, the ion pair dissociates long enough for the carbanion to turn over and recapture the proton:
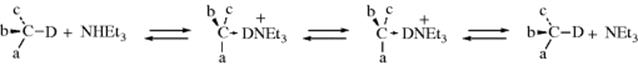
Thus, inversion (and hence racemization, which is produced by repeated acts of inversion) occurs without exchange. A single act of inversion without exchange is called isoinversion.
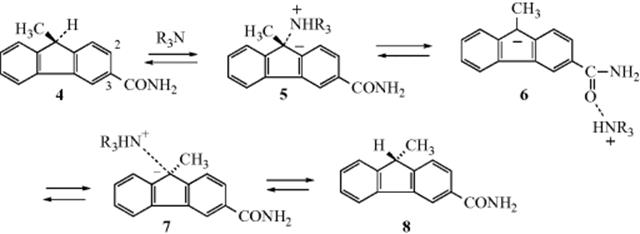
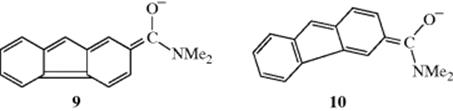
The isoinversion process can take place by a pathway in which a positive species migrates in a stepwise fashion around a molecule from one nucleophilic position to another. For example, in the exchange reaction of 3-carboxamido-9-methylfluorene (4) with Pr3N in t-BuOH, it has been proposed that the amine removes a proton from the 9 position of 4 and conducts the proton out to the C=O oxygen (6), around the molecule, and back to C-9 on the opposite face of the anion. Collapse of 7 gives the inverted product 8. Of course, 6 could also go back to 4, but a molecule that undergoes the total process 4 → 5 → 6 → 7 → 8 has experienced an inversion without an exchange. Evidence for this pathway, called the conducted tour mechanism,30 is that the 12-carboxamido isomer of 4 does not give isoracemization. In this case, the negative charge on the oxygen atom in the anion corresponding to 6 is less, because a canonical form in which oxygen acquires a full negative charge (9) results in disruption of the aromatic sextet in both benzene rings (cf. 10 where one benzene ring is intact). Whether the isoracemization process takes place by the conducted tour mechanism or a simple nonstructured contact ion–pair mechanism depends on the nature of the substrate (e.g., a proper functional group is necessary for the conducted tour mechanism) and of the base.31
It is known that vinylic carbanions can maintain configuration, so that SE1 mechanisms should produce retention, which is the case. For example, trans-2-bromo-2-butene was converted to 64–74% angelic acid:32
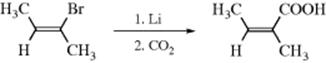
Only ~5% of the cis isomer (tiglic acid) was produced. In addition, certain carbanions in which the negative charge is stabilized by d-orbital overlap can maintain configuration (Sec. 5.B.ii) and SE1 reactions involving them proceed with retention of configuration.
12.A.iii. Electrophilic Substitution Accompanied by Double-Bond Shifts

When electrophilic substitution is carried out at an allylic substrate, the product may be rearranged (11 → 12). This type of process is analogous to the nucleophilic allylic rearrangements discussed in Section 10.D. There are two principal pathways. The first of these is analogous to the SE1 mechanism in that the leaving group is first removed, giving a resonance-stabilized allylic carbanion, which then attacks the electrophile Y.
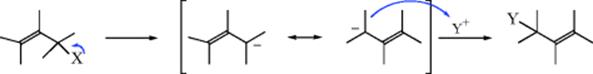
In the other pathway, the Y group is first attacked by the π-bond, giving a carbocation, which then loses X with formation of the alkene unit.
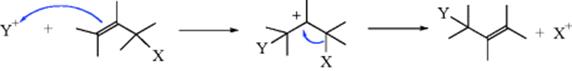
These mechanisms are more fully discussed under Reaction 12-2.
Most electrophilic allylic rearrangements involve loss of hydrogen, but they have also been observed with metallic leaving groups.33 Sleezer et al.34 found that crotylmercuric bromide reacted with HCl ~107 times faster than n-butylmercuric bromide and the product was >99% 1-butene. These facts point to an SEi′ mechanism (IUPAC designation cyclo-1/3/DEAEDnAn):
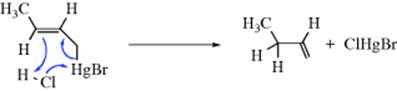
The reaction of the same compound with acetic acid–perchloric acid seems to proceed by an SE2′ mechanism (IUPAC designation 1/3/DEAE)34:
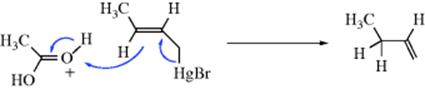
The geometry of electrophilic allylic rearrangement has not been studied very much (cf. the nucleophilic case, Sec. 10.E), but in most cases the rearrangement takes place with antistereoselectivity,35 although syn stereoselectivity has also been demonstrated.36 In one case, use of the electrophile H+ and the leaving group SnMe3 gave both syn and anti stereoselectivity, depending on whether the substrate was cis or trans.37
12.A.iv. Other Mechanisms
Addition–elimination (Reaction 12-16) and cyclic mechanisms (Reaction 12-40) are also known.
Much less work has been done on electrophilic aliphatic substitution mechanisms than on nucleophilic substitutions. The exact mechanisms of many of the reactions in this chapter are in doubt. For many of them, not enough work has been done to permit us to decide which of the mechanisms described in this chapter is operating, if indeed any is. There may be other electrophilic substitution mechanisms, and some of the reactions in this chapter may not even be electrophilic substitutions at all.