March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 13. Aromatic Substitution: Nucleophilic and Organometallic
In Section 10.G, category 2, it was pointed out that nucleophilic substitutions proceed so slowly at an aromatic carbon that the reactions of Chapter 10 are not feasible for aromatic substrates. There are, however, exceptions to this statement, and these exceptions form the subject of this chapter.1 Reactions that are successful at an aromatic substrate are largely of five kinds: (1) reactions activated by electron-withdrawing groups ortho and para to the leaving group; (2) reactions catalyzed by very strong bases and proceeding through aryne intermediates; (3) reactions initiated by electron donors; (4) reactions in which the nitrogen of a diazonium salt is replaced by a nucleophile; and (5) coupling reactions catalyzed by transition metals, primarily Pd,2 Cu, Ni, and so on. Note that solvent effects can be important.3 The transition metal catalyzed coupling reactions are included because they involve replacement of a leaving group on an aromatic ring.
13.A. Mechanisms
There are four principal mechanisms for aromatic nucleophilic substitution.4 Each of the four is similar to one of the aliphatic nucleophilic substitution mechanisms discussed in Chapter 10.
13.A.i. The SNAr Mechanism5
By far the most important mechanism for nucleophilic aromatic substitution consists of two steps, attack of the nucleophilic species at the ipso carbon of the aromatic ring (the carbon bearing the leaving group in this case), followed by elimination of the leaving group and regeneration of the aromatic ring.
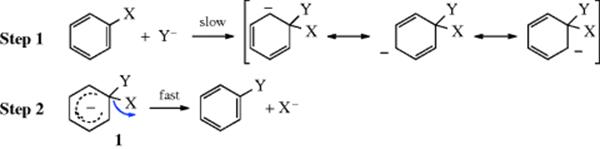
The first step is usually, but not always, rate determining. It can be seen that this mechanism greatly resembles the tetrahedral mechanism discussed in Chapter 16 and, in another way, the arenium ion mechanism of electrophilic aromatic substitution discussed in Chapter 11. In all three cases, the attacking species forms a bond with the substrate, giving an intermediate (e.g., 1) and then the leaving group departs: This mechanism is the SNAr mechanism.6The IUPAC designation is AN + DN (the same as for the tetrahedral mechanism; cf. the designation AE + DE for the arenium ion mechanism). This mechanism is generally found where activating groups are present on the ring (see Sec. 13.B.i).
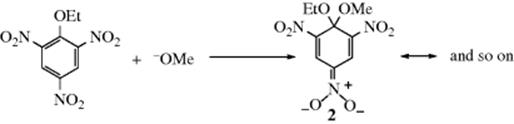
There is a great deal of evidence for the mechanism.4 Probably the most convincing evidence was the isolation, as long ago as 1902, of the intermediate 2 in the reaction between 2,4,6-trinitrophenetole and methoxide ion.7Intermediates of this type are stable salts, called Meisenheimer or Meisenheimer–Jackson salts,8 and many more have been isolated.9 The structures of several of these intermediates have been proved by NMR10 and by X-ray crystallography.11 Further evidence comes from studies of the effect of the leaving group on the reaction. If the mechanism were similar to either the SN1 or SN2 mechanisms described in Chapter 10, the Ar–X bond would be broken in the rate-determining step. In the SNAr mechanism, this bond is not broken until after the rate-determining step (i.e., if step 1 is rate determining). There is some evidence that electron transfer may be operative during this process.12 If the SNAr mechanism is operating, a change in leaving group should not have much effect on the reaction rate. In the reaction of dinitro compound 3 with piperidine, when X was Cl, Br, I, SOPh, SO2Ph, or p-nitrophenoxy, the rates differed only by a factor of ~5.13 This behavior would not be expected in a reaction in which the Ar–X bond is broken in the rate-determining step. The rates are not expected to be identical, because the nature of X affects the rate at which Y attacks.14 An increase in the electronegativity of X causes a decrease in the electron density at the site of attack, resulting in a faster attack by a nucleophile. Thus, in the reaction just mentioned, when X = F, the relative rate was 3300 (compared with I = 1). The very fact that fluoro is the best leaving group among the halogens in most aromatic nucleophilic substitutions is good evidence that the mechanism is different from the SN1 and the SN2 mechanisms, where fluoro is by far the poorest leaving group of the halogens. This is an example of the element effect (Sec. 10.F).

The pattern of base catalysis of reactions with amine nucleophiles provides additional evidence. Bases only catalyze these reactions when a relatively poor leaving group (e.g., OR) is present (not Cl or Br) and only when relatively bulky amines are nucleophiles.15 Bases could not catalyze step 1, but if amines are nucleophiles, bases can catalyze step 2. Base catalysis is found precisely in those cases where the amine moiety cleaves easily, but X does not, so that k1 is large and step 2 is rate determining. This is evidence for the SNAr mechanism because it implies two steps. Furthermore, in cases where bases are catalysts, they catalyze only at low-base concentrations: A plot of the rate against the base concentration shows that small increments of base rapidly increase the rate until a certain concentration of base is reached, after which further base addition no longer greatly affects the rate. This behavior, based on a partitioning effect (see Sec. 11.A.i), is also evidence for the SNAr mechanism. At low-base concentration, each increment of base, by increasing the rate of step 2, increases the fraction of intermediate that goes to product rather than reverting to reactants. At high-base concentration, the process is virtually complete: There is very little reversion to reactants and the rate becomes dependent on step 1. Just how bases catalyze step 2 has been investigated. For protic solvents, two proposals have been presented. One is that step 2 consists of two steps: rate-determining deprotonation of 4 followed by
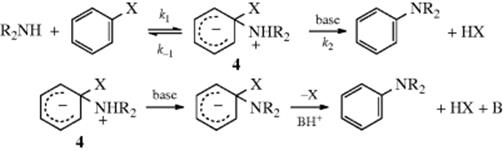
rapid loss of X, and that bases catalyze the reaction by increasing the rate of the deprotonation step.16 According to the other proposal, loss of X assisted by BH+ is rate determining.17 Two mechanisms, both based on kinetic evidence, have been proposed for aprotic solvents (e.g., benzene). In both proposals, the ordinary SNAr mechanism operates, but in one the attacking species involves two molecules of the amine (the dimer mechanism),18 while in the other there is a cyclic transition state.19 Further evidence for the SNAr mechanism has been obtained from ![]() and
and ![]() isotope effects.20
isotope effects.20
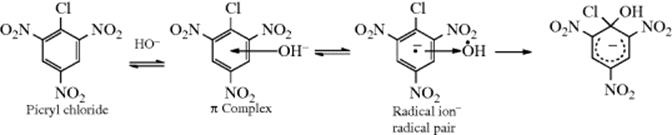
Step 1 of the SNAr mechanism has been studied for the reaction between picryl chloride (as well as other substrates) and ![]() ions (Reaction 13-1), and spectral evidence has been reported21 for two intermediates, one a π complex (Sec. 11.A.i), and the other a radical ion–radical pair.
ions (Reaction 13-1), and spectral evidence has been reported21 for two intermediates, one a π complex (Sec. 11.A.i), and the other a radical ion–radical pair.
As with the tetrahedral mechanism at an acyl carbon, nucleophilic catalysis (Sec. 16.A.i) has been demonstrated with an aryl substrate, in certain cases.22 There is also evidence of an interaction of anions with the π-cloud of aromatic compounds.23
13.A.ii. The SN1 Mechanism
For aryl halides and sulfonates, even active ones, a unimolecular SN1 mechanism (IUPAC: DN + AN) is very rare; it has only been observed for aryl triflates in which both ortho positions contain bulky groups (tert-butyl or SiR3).24 It is in reactions with diazonium salts25 that this mechanism is important26:
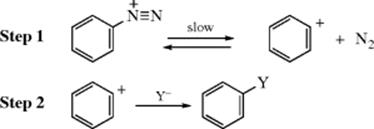
Among the evidence for the SN1 mechanism27 with aryl cations as intermediates,28,29 is the following:30
1. The reaction rate is first order in diazonium salt and independent of the concentration of Y.
2. When high concentrations of halide salts are added, the product is an aryl halide, but the rate is independent of the concentration of the added salts.
3. The effects of ring substituents on the rate are consistent with a unimolecular rate-determining cleavage.31
4. When reactions were run with substrate deuterated in the ortho position, isotope effects of ~1.22 were obtained.32 It is difficult to account for such high secondary isotope effects in any other way except that an incipient phenyl cation is stabilized by hyperconjugation (see Sec. 2.M),33 which is diminished when hydrogen is replaced by deuterium.
![]()
5. That the first step is reversible cleavage34 was demonstrated by the observation that when ![]() was the reaction species, recovered starting material contained not only
was the reaction species, recovered starting material contained not only ![]() , but also
, but also ![]() .35,36 This could arise only if the nitrogen breaks away from the ring and then returns. Additional evidence was obtained by treating
.35,36 This could arise only if the nitrogen breaks away from the ring and then returns. Additional evidence was obtained by treating ![]() with unlabeled N2 at various pressures. At 300 atm, the recovered product had lost ~3% of the labeled nitrogen, indicating that PhN2+ was exchanging with atmospheric N2.36
with unlabeled N2 at various pressures. At 300 atm, the recovered product had lost ~3% of the labeled nitrogen, indicating that PhN2+ was exchanging with atmospheric N2.36
There is kinetic and other evidence37 that step 1 is more complicated and involves two steps, both reversible:
![]()
Intermediate 5, which is probably some kind of a tight ion–molecule pair, has been trapped with carbon monoxide.38
13.A.iii. The Benzyne Mechanism39
Some aromatic nucleophilic substitutions are clearly different in character from those that occur by the SNAr mechanism (or the SN1 mechanism). These substitutions occur with aryl halides that have no activating groups; stronger bases are required than those normally used; and the incoming group does not always take the position vacated by the leaving group. The validity of the latter statement was elegantly demonstrated by the reaction of 1-![]() -chlorobenzene with potassium amide:
-chlorobenzene with potassium amide:
![]()
The product consisted of almost equal amounts of aniline labeled in the 1 and 2 positions.40
A mechanism that can explain all these facts involves elimination followed by addition. In step 1, a suitable base removes the ortho hydrogen, with subsequent (or concomitant) loss of the chlorine (leaving group) to
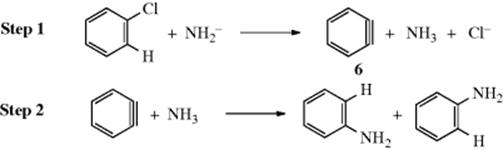
generate symmetrical intermediate 641 called benzyne42 (see below).43 In step 2, benzyne is attacked by the NH3 at either of two positions, which explains why about one-half of the aniline produced from the radioactive chlorobenzene was labeled at the 2 position. The fact that the 1 and 2 positions were not labeled equally is the result of a small isotope effect. Other evidence for this mechanism follows:
1. If the aryl halide contains two ortho substituents, the reaction should not be able to occur. This is indeed the case.38
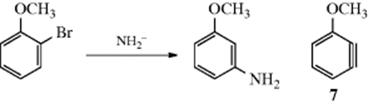
2. It had been known many years earlier that aromatic nucleophilic substitution occasionally results in substitution at a different position. This is called cine substitution44 and can be illustrated by the conversion of o-bromoanisole to m-aminoanisole.45 In this particular case, only the meta isomer is formed. The reason a 1:1 mixture is not formed is that the intermediate 7 is not symmetrical and the methoxy group directs the incoming group meta, but not ortho (see Sec. 13.B.i). However, not all cine substitutions proceed by this kind of mechanism (see Reaction 13-30). A study of the influence of structure on the formation of substituted benzynes, along with rate studies, has been reported.46
3. The fact that the order of halide reactivity is Br > I > Cl > F (when the reaction is performed with KNH2 in liquid NH3) shows that the SNAr mechanism is not operating here.40
In the conversion of the substrate to 7, either proton removal or subsequent loss of halide ion can be rate determining. In fact, the unusual leaving-group order just mentioned (Br > I > Cl) stems from a change in the rate-determining step. When the leaving group is Br or I, proton removal is rate determining and the rate order for this step is F > Cl > Br > I. When Cl or F is the leaving group, cleavage of the C–X bond is rate determining and the order for this step is I > Br > Cl > F. Confirmation of the latter order was found in a direct competitive study. meta-Dihalobenzenes in which the two halogens are different were treated with ![]() .47 In such compounds, the most acidic hydrogen is the one between the two halogens; when it leaves, the remaining anion can lose either halogen. Therefore a study of which halogen is preferentially lost provides a direct measure of leaving-group ability. The order was found to be I > Br > Cl.47,48
.47 In such compounds, the most acidic hydrogen is the one between the two halogens; when it leaves, the remaining anion can lose either halogen. Therefore a study of which halogen is preferentially lost provides a direct measure of leaving-group ability. The order was found to be I > Br > Cl.47,48
Species, such as 6 and 7, are called benzynes (sometimes dehydrobenzenes), or more generally, arynes.49 The mechanism in which such species are intermediates is known as the benzyne mechanism. Benzynes are very reactive, and neither benzyne nor any other aryne has yet been isolated under ordinary conditions,50 but benzyne has been isolated in an Ar matrix at 8 K,51 and its IR spectrum is observed. In addition, benzynes can be trapped (e.g., they undergo the Diels–Alder reaction, See Reaction 15-60). Note that the extra pair of electrons does not affect the aromaticity. However, evaluation by a series of aromaticity indicators, including magnetic susceptibility anisotropies and exaltations, nucleus-independent chemical shifts (NICS), aromatic stabilization energies, and valence bond Pauling resonance energies point to the o-benzyne > m-benzyne > p-benzyne aromaticity order.52 The relative order with respect to benzene depends on the aromaticity criterion.48 Note that tunable reactivity for m-benzynes has been demonstrated.53 The aromatic sextet from the aromatic precursor functions as a closed ring, and the two additional electrons are merely located in a π orbital that covers only two carbons. Benzynes do not have a formal triple bond, since two canonical forms (A and B) contribute to the hybrid. The Ir spectrum, mentioned above, indicates that Acontributes more than B. Not only benzene rings, but also other aromatic rings54 and even non-aromatic rings (Sec. 10.F) can react through this kind of intermediate. Of course, the non-aromatic rings do have a formal triple bond. When a benzyne unit is fused to a small ring, strain induced regioselectivity was observed in its reactions.55
13.A.iv. The SRN1 Mechanism
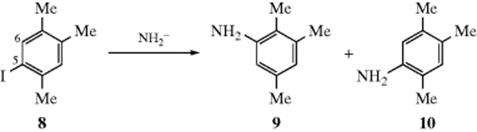
When 5-iodo-1,2,4-trimethylbenzene (8) was treated with KNH2 in NH3, 9 and 10 were formed in the ratio 0.63:1. The presence of an unactivated substrate, a strong base, and the occurrence of cine substitution along with normal substitution are strong indications of a benzyne mechanism. Yet if that were so, the 6-iodo isomer of 8 should have given 9 and 10 in the same ratio (because the same aryne intermediate would be formed in both cases), but in this case the ratio of 9 to 10 was 5.9:1 (the chloro and bromo analogues did give the same ratio (1.46:1), showing that the benzyne mechanism may be taking place there).
To explain the iodo result, it has been proposed56 that in addition to the benzyne mechanism, a free radical mechanism is also operating here:
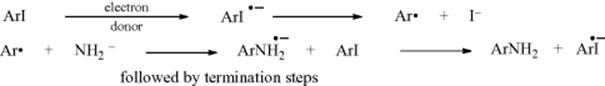
This is called the SRN1 mechanism,57 and many other examples are known (see Reactions 13-3, 13-4, 13-6, and 13-14). The IUPAC designation is T + DN + AN.58 Note that the last step of the mechanism produces ArI√− radical ions, so the process is a chain mechanism59 (see Sec. 14.A.i). An electron donor is required to initiate the reaction. In the case above, it was solvated electrons from KNH2 in NH3. Evidence was that the addition of potassium metal (a good producer of solvated electrons in ammonia) completely suppressed the cine substitution. Further evidence for the SRN1 mechanism was that addition of radical scavengers, which would suppress a free radical mechanism, led to 9:10 ratios much closer to 1.46:1. Numerous other observations of SRN1 mechanisms that were stimulated by solvated electrons and inhibited by radical scavengers have also been recorded.60 Further evidence for the SRN1 mechanism in the case above was that some 1,2,4-trimethylbenzene was found among the products. This could easily be formed by abstraction by Ar√ of H from the solvent NH3. Besides initiation by solvated electrons,61 SRN1 reactions have been initiated photochemically,62 electrochemically,63 and even thermally.64
The SRN1 reactions have a fairly wide scope. The efficiency of the reaction has been traced to the energy level of the radical anion of the substitution product.65 There is no requirement for activating groups or strong bases, but in DMSO haloarenes are less reactive as the stability of the anion increases.66 The reaction has also been done in liquid ammonia, promoted by ultrasound (Sec. 7.B),67 and ferrous ion has been used as a catalyst.68 Alkyl, alkoxy, aryl, and COO− groups do not interfere, although Me2N, O−, and NO2 groups do interfere. Cine substitution is not found.
13.A.v. Other Mechanisms
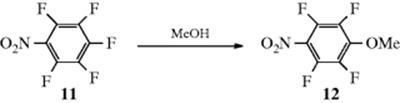
There is no clear-cut proof that a one-step SN2 mechanism, so important at a saturated carbon, ever actually occurs with an aromatic substrate. The hypothetical aromatic SN2 process is sometimes called the one-stage mechanism to distinguish it from the two-stage SNAr mechanism. A “clean” example of a SRN2 reaction has been reported, the conversion of 11 to 12 in methanol.69 Both SRN1 and SRN2 reactions have been reviewed.70
Some of the reactions in this chapter operate by still other mechanisms, among them an addition–elimination mechanism (see Reaction 13-17). A new mechanism has been reported in aromatic chemistry, a reductively activated “polar” nucleophilic aromatic substitution.71 The reaction of phenoxide with p-dinitrobenzene in DMF shows radical features that cannot be attributed to a radical anion, and it is not SRN2. The new designation was proposed to account for these results.