Organic Chemistry I For Dummies, 2nd Edition (2014)
Part II. Hydrocarbons
Chapter 9. Seeing Double: The Alkenes
IN THIS CHAPTER
Seeing the importance of alkenes
Determining the degrees of unsaturation
Adding alkene nomenclature to your repertoire
Defining alkene stereochemistry
At this point, I assume you know the basics of organic chemistry. I assume you understand the difference between covalent and ionic bonds (Chapter 2), how to draw and interpret organic structures (Chapter 3), and how to name alkanes (Chapter 7). In construction terms, I assume you have the foundations of the house of organic chemistry. Now that the foundation is there, I get to the fun stuff — the chemical reactions, themselves — which build up the walls of the house of organic chemistry (that Jack built).
Specific chemical reactions are the meat and potatoes of most organic chemistry courses. And as the number of reactions start to pile up like dirty socks on the floors of college dormitories, organizing all the many reactions in as succinct and convenient a way as possible becomes essential. With the alkene functional group, numerous reactions are available to form alkenes and convert them into other functional groups.
In this chapter, I introduce the alkene functional group. I show you how to add the nomenclature of alkenes into your repertoire, and I discuss the many reactions that both make alkenes and transform them into new kinds of compounds. Along the way, I give you practical advice on how you can best learn the swarms of reactions that you see throughout this chapter and in your organic course.
Defining Alkenes
Alkenes are compounds that contain carbon-carbon double bonds. Because alkenes are so often found in valuable compounds (like pharmaceuticals), they’re one of the most important functional groups in organic chemistry. Alkenes are also very versatile, because they’re easy to make and convert into other molecules, as Figure 9-1 shows. Therefore, alkenes become useful as waypoints on the road to synthesizing other molecules.
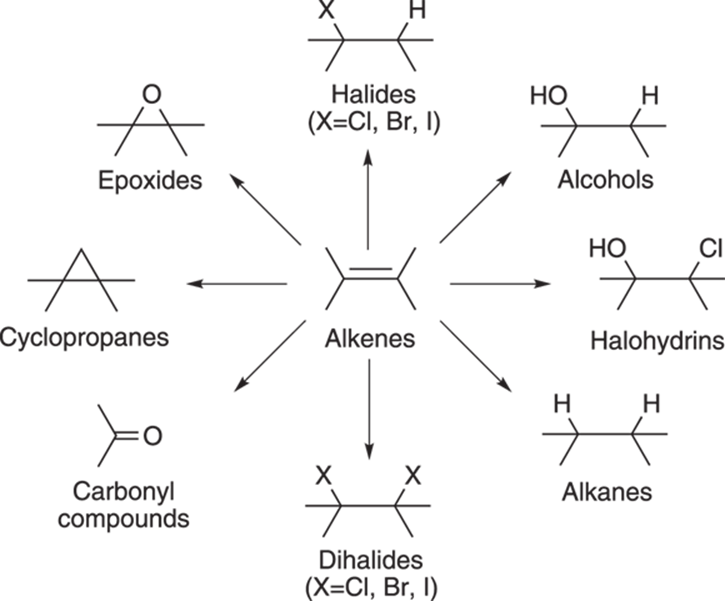
FIGURE 9-1: Some of the compounds that can be made from alkenes.
What do I mean by waypoints? Think of air travel as an analogy. You may not be able to get a flight directly from Tampa to Timbuktu, but you may be able to get a connection flight from Tampa to Chicago, and from Chicago you could get a flight to Timbuktu. Similarly, you may not be able to convert one functional group directly into the functional group that you want, but you may be able to convert the functional group into a connection functional group (like an alkene), which could then be converted into the desired product. So, even if a desired product contains no carbon-carbon double bonds, alkenes may still play a role in the synthesis of that molecule.
MASTERING ORGANIC REACTIONS
Many students find that making reaction note cards (like the one shown in the accompanying figure) is a helpful way to learn the reactions (I recommend this practice). Other students find that reaction schemes that organize functional group conversions visually (like the one shown in Figure 9-1) are very helpful for learning the reactions.
No matter how you choose to learn the reactions, you should work many problems that test your ability to recall reactions and apply these reactions to new situations. Because most of organic chemistry involves reactions of organic molecules, you need to become an “expert” in synthetic organic chemistry. And to become an expert at anything you need to practice. To become an expert pianist, you need to practice the piano; to become an expert painter, you need to practice painting; and to become an expert whistler, you need to practice whistling. In the same way, to become an expert in organic reactions, you need to practice, practice, practice.
In a practical sense, you should work many problems that test both your ability to recall the reactions (including which reagents to use), and your ability to apply what you know to new circumstances. At first, you’ll just deal with one-step reactions — what reagents convert compound A into compound B, for example. But eventually you’ll need to be able to work multistep synthesis problems — that is, syntheses involving more than one step, which I discuss in more detail in Appendix A.
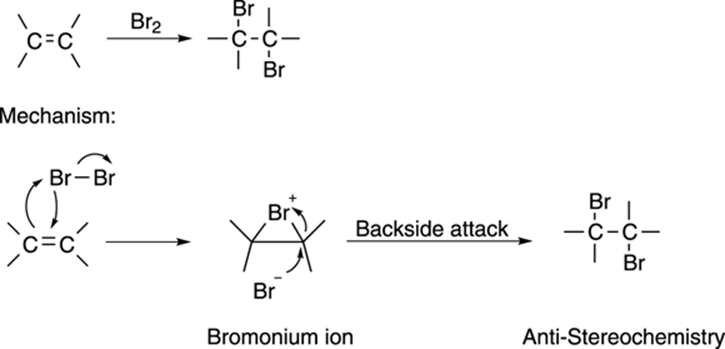
Taking Away Hydrogens: Degrees of Unsaturation
Alkanes are said to be saturated hydrocarbons because these molecules are saturated with hydrogens, holding the maximum number of hydrogens while still obeying all the rules of valence (having no more than four bonds for second-row atoms). Because adding a carbon-carbon double bond in a molecule requires the loss of two hydrogens, alkenes are said to add a degree of unsaturation into a molecule (sometimes called a double-bond equivalent or index of hydrogen deficiency). So, although alkanes have the general formula CnH2n+2, where n is the number of carbon atoms in the molecule, alkenes have the general formula of CnH2n, and contain two fewer hydrogens than an alkane with the same number of carbons.
Determining degrees of unsaturation from a structure
Knowing the number of degrees of unsaturation in a molecule is useful because this number gives you an indication of how many double bonds are present in an unknown compound. (This morsel of information becomes very useful when you want to determine the structure of an unknown compound. See Part 4 for how to apply this information to spectroscopy and spectrometry problems.)
The degrees of unsaturation in a molecule are additive — a molecule with one double bond has one degree of unsaturation, a molecule with two double bonds has two degrees of unsaturation, and so forth. Just as the formation of a double bond causes two hydrogens to be lost, the formation of a ring also results in the loss of two hydrogens, so every ring in the molecule also adds one degree of unsaturation. For every triple bond, two degrees of unsaturation are added to a molecule, because a molecule must lose four hydrogens to make a triple bond. Some examples of three-carbon molecules with different numbers of degrees of unsaturation are shown in Figure 9-2.
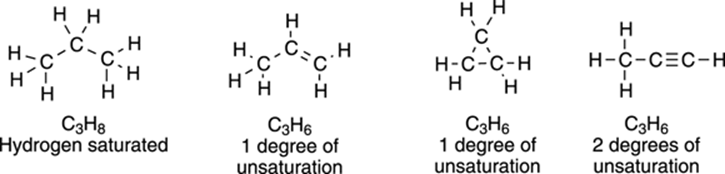
FIGURE 9-2: The degrees of unsaturation for three-carbon molecules.
To determine the number of degrees of unsaturation for any arbitrary structure, you sum all the individual elements of unsaturation in the molecule. Figure 9-3 shows a molecule that consists of one ring, one double bond, and one triple bond. This molecule, therefore, has four degrees of unsaturation because the double bond and the ring each add one degree of unsaturation, and the triple bond adds two degrees, for a total of four.
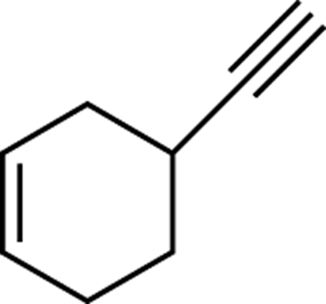
FIGURE 9-3: A molecule with four degrees of unsaturation.
Problem solving: Determining degrees of unsaturation from a molecular formula
More important than determining the number of degrees of unsaturation from a molecular structure is being able to determine the number of degrees of unsaturation from a molecular formula. The number of degrees of unsaturation can be determined from the molecular formula using the following equation.
![]()
With this equation, the number of degrees of unsaturation can be determined for any hydrocarbon whose molecular formula is known. (For compounds whose structure and formula are not known, chemists use an instrumental technique called mass spectrometry to determine the molecular formula of the compound. See Chapter 17 for more on mass spec.)
But what about molecules that contain atoms other than hydrogen and carbon? In such cases, you need to convert these multi-atom molecular formulas into equivalent formulas that contain just carbon and hydrogen so they can be plugged into the preceding equation. To do so, you use the following conversion factors:
· Halogens ( F, Cl, Br, I ): Add one hydrogen to the molecular formula for each halogen present.
· Nitrogen: Subtract one hydrogen for each nitrogen present.
· Oxygen or sulfur: Ignore.
For example, to determine the number of degrees of unsaturation in the formula C8H6F3NO2, you first make the proper substitutions for all atoms that are not hydrogen and carbon. Fluorine is a halogen, so you add three hydrogen atoms to the molecular formula (one for each F). The molecule contains one nitrogen, so you subtract one hydrogen from the molecular formula. The two oxygens in the molecule you ignore. This gives a reduced equation of C8H6+3–1 = C8H8. In other words, both the formula C8H6F3NO2 and the formula C8H8 have identical numbers of degrees of unsaturation. Plugging this reduced formula into the preceding equation gives five degrees of unsaturation for the molecular formula C8H6F3NO2.
The Nomenclature of Alkenes
If you know how to name alkanes (see Chapter 7), adding alkene nomenclature to your repertoire is a fairly straightforward task. Whereas the names of alkanes end with the suffix –ane, alkenes end with the suffix –ene. A two-carbon alkene, therefore, is named ethene; a three-carbon alkene is named propene; and an alkene in a five-membered ring is named cyclopentene (see Figure 9-4).
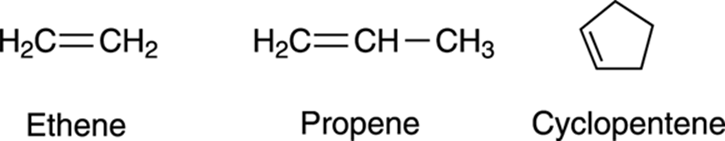
FIGURE 9-4: The structures of some alkenes.
Numbering the parent chain
For molecules in which the double bond can be put in more than one position, you must put a number in front of the parent name to indicate the position of the double bond. For example, two alkene locations in pentene are possible (see Figure 9-5), so a number must be placed in front of the name to show the location of the double bond.

FIGURE 9-5: The two possible locations.
The parent chain is numbered so that the alkene is given the lowest possible number. For example, you wouldn’t name the pentene 3-pentene because that would be an identical structure to 2-pentene. Figure 9-6 shows the correct and incorrect numbering in a seven-carbon chain with a methyl substituent. Notice that giving the smaller number to the double bond takes precedence over assigning the smaller number to the substituent.
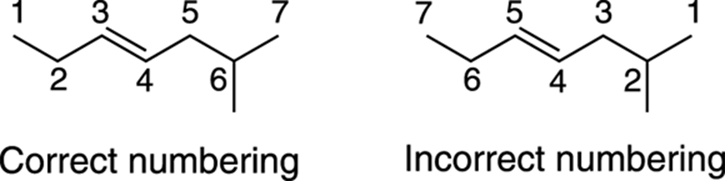
FIGURE 9-6: The correct and incorrect numberings of a long-chain alkene.
Additionally, the alkene must be included in the parent chain, even if a longer chain of carbon atoms can be found that doesn’t include the alkene. Figure 9-7 shows how this works.
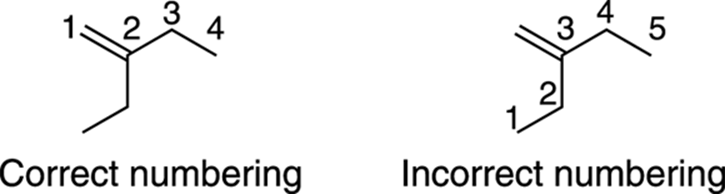
FIGURE 9-7: The correct and incorrect numberings of an alkene.
To number an alkene in a ring, number around the ring so that the double bond has the lowest possible number, as shown in Figure 9-8.

FIGURE 9-8: The correct and incorrect numberings of a ringed alkene.
Adding multiple double bonds
The naming of alkenes with more than one double bond requires the use of a prefix (such as di–, tri–, tetra–, and so forth) to indicate the number of double bonds in the molecule. Also, the position of all double bonds is indicated with numbers at the beginning of the name. An example of how this is done is shown in Figure 9-9.
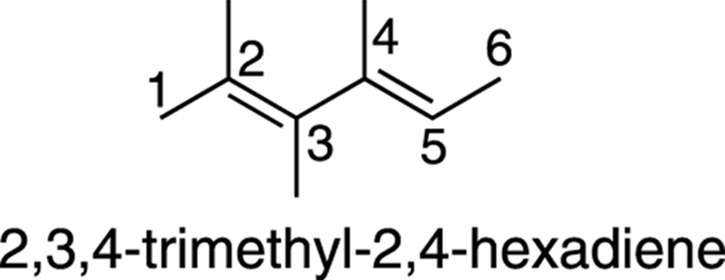
FIGURE 9-9: The numbering and naming of a dialkene containing two methyl substituents.
Common names of alkenes
Although alkenes can be named systematically using the IUPAC nomenclature (the official nomenclature system of chemists) as outlined earlier, some alkenes have common names that come from the olden days. These older names (some of which are shown in Figure 9-10) are still often seen in the literature and need to be memorized. For some smaller alkenes, the suffix –ylene is used instead of simply –ene. Ethene, for example, is usually called ethylene, and propene is called propylene. Styrene — the compound used to make the plastic polystyrene — is the common name for a molecule that consists of a double bond jutting off a benzene ring (see Chapter 15 for a discussion of benzene and other ringed compounds).
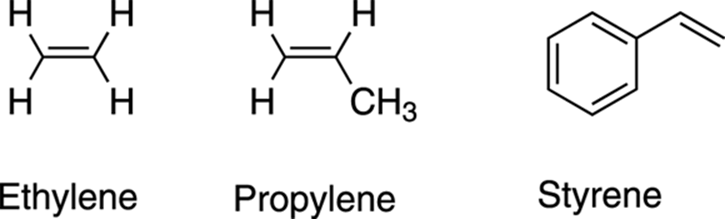
FIGURE 9-10: The common names of some alkenes.
The Stereochemistry of Alkenes
Unlike carbon-carbon single bonds, which are free to rotate (see Chapter 8), double bonds are fixed and rigid. In other words, rotation around carbon-carbon double bonds is not possible at reasonable temperatures. Therefore, molecules containing double bonds have the possibility of having stereoisomers, just as ring systems do.
 Molecules that have the same connectivity of atoms, but different orientations of those atoms in space are called stereoisomers (see Chapter 6).
Molecules that have the same connectivity of atoms, but different orientations of those atoms in space are called stereoisomers (see Chapter 6).
You on my side or their side? Cis and trans stereochemistry
Consider the case of 2-pentene, shown in Figure 9-11. In this alkene, two stereoisomers are possible. One stereoisomer, called the cis stereoisomer, has both of the double-bond hydrogens on the same side of the double bond, while the other stereoisomer, called the trans stereoisomer, has the two hydrogens on opposite sides of the double bond. In general, when two identical groups are on the same side of the double bond, the molecule is said to possess cis stereochemistry; when two identical groups are on opposite sides of the double bond, the molecule is said to possesstrans stereochemistry.
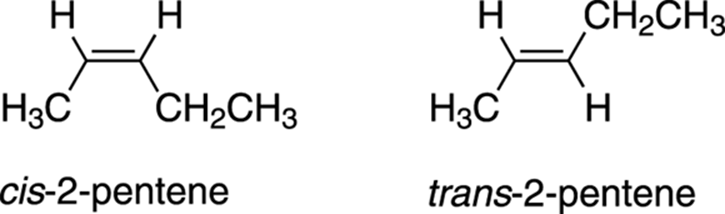
FIGURE 9-11: Cis and trans 2-pentene.
You can only have cis-trans stereochemistry in rings and on double bonds. You can’t have cis-trans isomers on single bonds due to the rapid free rotation of these bonds at room temperature. (Take a peek at the section on Newman projections and conformations in Chapter 8.)
Playing a game of high-low: E/Z stereochemistry
What if a double bond holds four nonidentical groups? In such a case, the cis-trans nomenclature doesn’t apply because cis-trans nomenclature can be used only when two identical groups are attached to a double bond or ring (in many cases, these identical groups are simply hydrogens). When four nonidentical groups are attached to a double bond, you must use the E/Z system of nomenclature (rather than the cis-trans nomenclature) to assign the stereochemistry of the double bond. (E stands for the German word entgegen, which means “opposite,” while Z stands for the German word zusammen, which means “together.”)
To use the E/Z system of nomenclature, you have to play a game of high-low. First, you must decide which substituent on each carbon is given higher priority and which is given lower priority using the Cahn–Ingold–Prelog prioritizing scheme on both sides of the alkene. Figure 9-12helps you visualize this process.
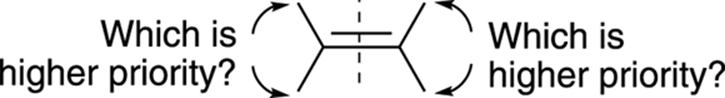
FIGURE 9-12: Assigning E/Z stereochemistry.
If the two high-priority substituents are on the same side of the double bond, the alkene is given the Z nomenclature; if the highs are on opposite sides of the double bond, the alkene is given the E nomenclature. (See Figure 9-13 for an illustration of these nomenclature rules.)

FIGURE 9-13: Playing high-low.
Here’s how to prioritize the double-bond substituents using the Cahn–Ingold–Prelog prioritizing scheme.
· Individual substituents: The substituent whose first atom has the highest atomic number gets the highest priority. For example, iodine would be higher priority than bromine, which would be higher than fluorine.
· Ties: In the case of a tie (both first atoms are carbon, for example), proceed to the next atom and decide which atom has the higher atomic number. If you get another tie, keep going until the tie is broken. (See Figure 9-14 for an example.)
· Multiple bonds: You treat multiple bonds in a somewhat strange fashion using the Cahn–Ingold–Prelog prioritizing scheme. Using this prioritizing scheme, you treat multiple bonds as if they were multiple single bonds. A carbon double-bonded to oxygen, for example, would be treated as if the carbon had two carbon-oxygen single bonds (and as if each oxygen were bonded back to carbon). Which is higher priority, the carbon double-bonded to oxygen or the carbon triple-bonded to nitrogen, as shown in Figure 9-15? The carbon double-bonded to oxygen is higher priority because oxygen has a higher atomic number than nitrogen. (The fact that one group has three C-N bonds and the other has only two C-O bonds is irrelevant; the number of bonds only matters when comparing identical bonds, so C=NH would be higher priority than C-NH2.)
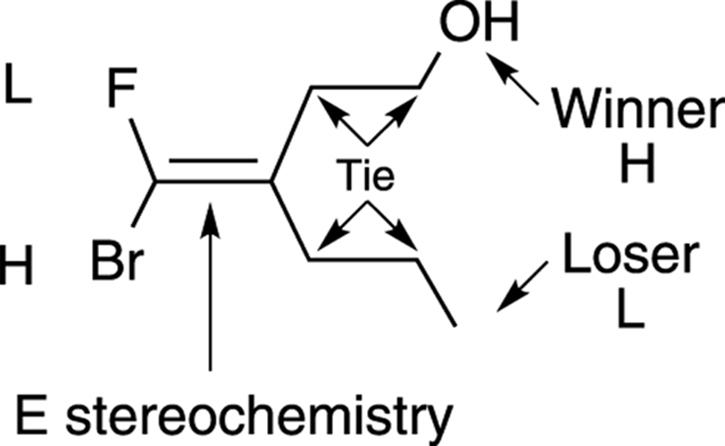
FIGURE 9-14: Determining the priorities of double-bond substituents in cases of ties.
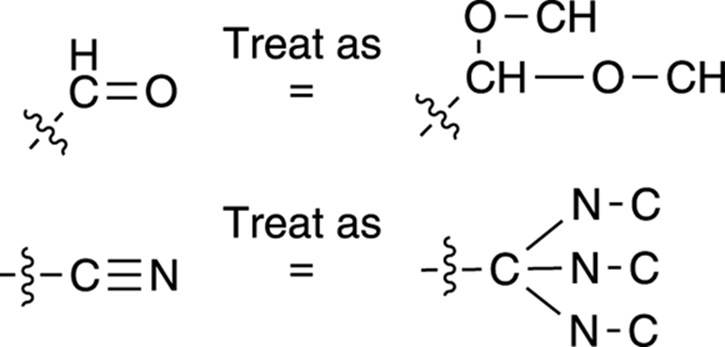
FIGURE 9-15: Treating multiple bonds using the Cahn–Ingold–Prelog prioritizing scheme.
Stabilities of Alkenes
Reactions yielding alkenes often preferentially form certain alkene isomers over others. In many cases, this preference for one isomer over another is the result of one isomer being more stable than another, with the more stable isomer being formed preferentially. Understanding what structural features lead to this greater alkene stability helps you to justify why certain alkenes will be formed in particular reactions.
Alkene substitution
Perhaps the biggest factor affecting the stability of alkenes is the number of non-hydrogen substituents that come off the carbon-carbon double bond. In general, the more non-hydrogen substituents that come off a double bond, the more stable the alkene is, as shown in Figure 9-16.
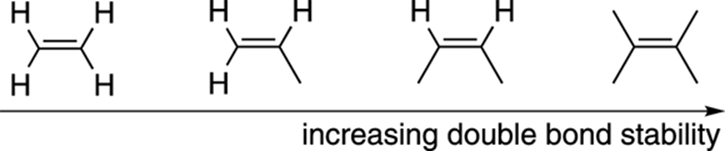
FIGURE 9-16: The relative stabilities of substituted alkenes.
Stability of cis and trans isomers
Also affecting the stability of alkenes is cis-trans isomerism. Generally, trans isomers of alkenes are more stable than cis isomers. This preference of alkenes for trans isomers comes about because atoms (like people) want a certain amount of personal space and don’t like to have their personal space invaded. Technically, this desire for adequate space results from atoms repelling each other because of the electron-electron repulsion of their electron clouds.
Alkenes in the trans configuration have substituents that are on opposite sides of the double bond, and the bulky substituents are, thus, farther away from each other; alkenes in the cis configuration, on the other hand, have substituents that are on the same side of the double bond, and the substituents are close to each other, invading each other’s space. The electron-electron repulsion that results from atoms being forced into the personal space of other atoms is called steric repulsion, as shown in Figure 9-17. Therefore, alkenes prefer to be in trans configuration rather thancis configuration.
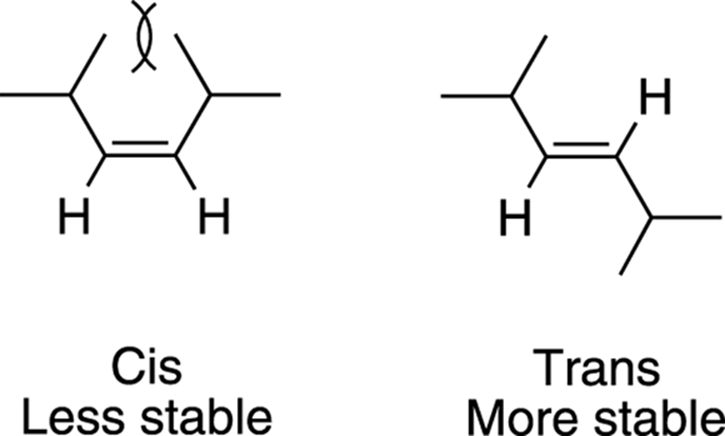
FIGURE 9-17: The steric repulsion of cis alkenes and the relative stabilities of cis and trans alkenes.
 To say that one molecule is more stable than another is generally synonymous with saying that one is lower in energy than another. In other words, stable molecules are generally lower in energy than unstable molecules.
To say that one molecule is more stable than another is generally synonymous with saying that one is lower in energy than another. In other words, stable molecules are generally lower in energy than unstable molecules.
Formation of Alkenes
The three primary ways to form alkenes is by dehydrohalogenation, dehydration, and the Wittig reaction.
Elimination of acid: Dehydrohalogenation
One of the most common methods of making alkenes is from alkyl halides. Halogens are often abbreviated X, which stands for any of the halogens chlorine (Cl), bromine (Br), or iodine (I). When a strong base (abbreviated B:–) is added to an alkyl halide, an elimination reaction occurs to make the alkene. This process is called dehydrohalogenation and is shown in Figure 9-18, indicating the loss of a hydrogen atom and a halide from the starting material in the reaction. The proton that’s eliminated is a proton adjacent to the halide, not a proton on the same carbon as the halide. The two possible mechanisms for this elimination are discussed in detail in Chapter 12.
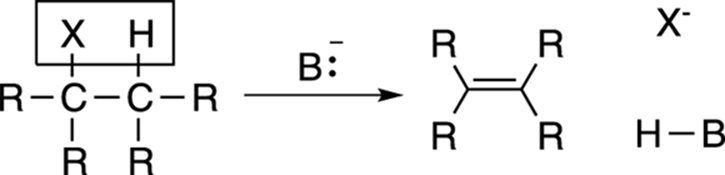
FIGURE 9-18: The dehydrohalogenation of an alkyl halide.
Losing water: Dehydration of alcohols
Similar to the process of dehydrohalogenation of alkyl halides is the process of dehydration, or the loss of water to make alkenes. In the presence of a strong acid and heat (abbreviated Δ), alcohols lose water to make alkenes, as shown in Figure 9-19. These dehydration reactions typically follow the E1 mechanism discussed in Chapter 12.
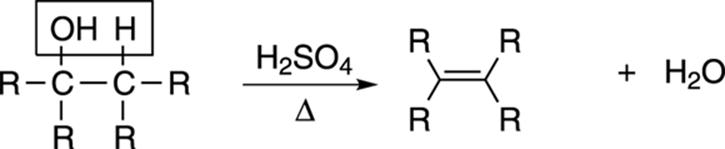
FIGURE 9-19: The dehydration of an alcohol.
Alkenes from coupling: The Wittig reaction
A very useful reaction for making alkenes is the Wittig reaction (pronounced vit-ig), a reaction for which Georg Wittig, the chemist who discovered it, received the Nobel Prize in chemistry.
 If you want to win a Nobel Prize in chemistry, a good way to start is to discover a general carbon-carbon bond-making reaction, because such reactions are extremely valuable. Chemists who have won Nobel Prizes for doing just that include Otto Diels, Kurt Alder, Georg Wittig, and Victor Grignard.
If you want to win a Nobel Prize in chemistry, a good way to start is to discover a general carbon-carbon bond-making reaction, because such reactions are extremely valuable. Chemists who have won Nobel Prizes for doing just that include Otto Diels, Kurt Alder, Georg Wittig, and Victor Grignard.
In the Wittig reaction, an aldehyde or ketone is reacted with a phosphorane (phosphorous double bonded to carbon, Ph3P=CR2) to make an alkene, as shown in Figure 9-20. (Remember that Ph stands for a phenyl ring, C6H5.) From a conceptual standpoint, you can imagine snipping off the carbonyl oxygen and the triphenylphosphine (PPh3) portion of the phosphorane, and then smooshing the two parts together to make the alkene.
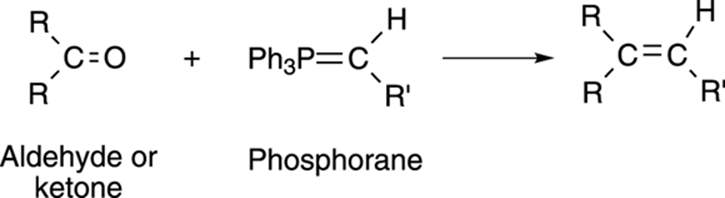
FIGURE 9-20: The Wittig reaction.
Making a phosphorane requires two steps. First, triphenylphosphine (Ph3P) is reacted with a primary alkyl halide to make a phosphonium ion, as shown in Figure 9-21. Then, a strong base (B:–) is added to deprotonate the carbon adjacent to the phosphorous, making the phosphorane; this phosphorane has two major resonance structures.
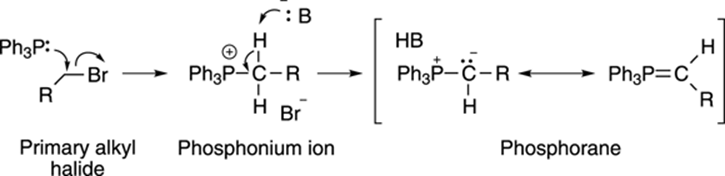
FIGURE 9-21: Making phosphorane.
The mechanism of the addition of the phosphorane to the carbonyl compound (usually an aldehyde or ketone) is still disputed, but chemists think that the attack of the phosphorane on the carbonyl compound generates a charged species called a betaine, as shown in Figure 9-22. Theoxyanion (negatively charged oxygen) of the betaine then attacks the positively charged phosphorous to generate a four-membered neutral species called an oxaphosphetane. The oxaphosphetane collapses to generate a very stable triphenylphosphine oxide and the alkene. (Whew!)
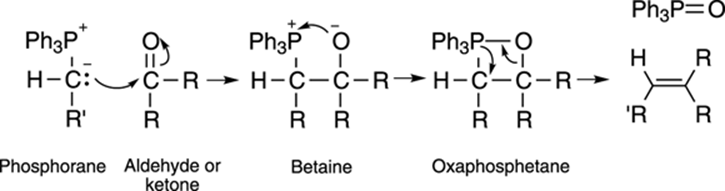
FIGURE 9-22: The mechanism of the Wittig reaction.