Organic Chemistry I For Dummies, 2nd Edition (2014)
Part II. Hydrocarbons
Chapter 10. Reactions of Alkenes
IN THIS CHAPTER
Adding a hydrohalic acid to a double bond
Clueing in on carbocations
Making alcohol by adding water across a double bond
Cleaving carbon-carbon double bonds into two fragments
Converting alkenes into cyclopropane rings
Sussing out the Simmons–Smith reaction
Making epoxides by reacting an alkene with a peroxy acid
Converting alkenes into alkanes by hydrogenation
One of the most useful aspects of alkenes is that they are incredibly versatile, as they can be easily made into such a variety of different compounds. In this chapter, I show you how you can convert alkenes into a variety of different functional groups like alkyl halides, alcohols, aldehydes and ketones, epoxides, and cyclopropane rings.
Adding Hydrohalic Acids across Double Bonds
Adding a hydrohalic acid (usually HCl or HBr) to a double bond converts the alkene into an alkyl halide. This reaction, shown in Figure 10-1, is just the reverse reaction of the elimination reaction that creates double bonds.
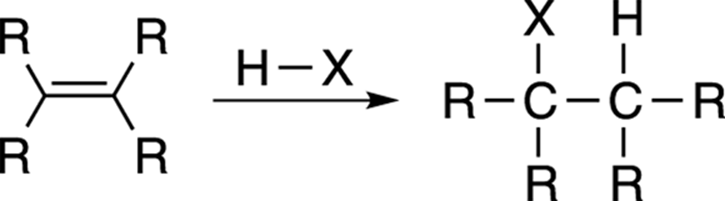
FIGURE 10-1: The Markovnikov addition to alkenes.
The mechanism of the reaction is shown in Figure 10-2. The first step of the reaction is protonation of the double bond by the acid. This leads to a short-lived carbocation intermediate (cations on carbon atoms are called carbocations). The carbocation is then attacked by the free halide anion to generate the alkyl halide.
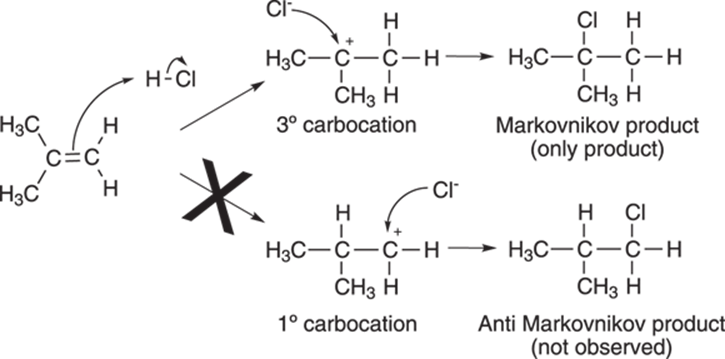
FIGURE 10-2: The formation of the carbocation on the most highly substituted carbon (the Markovnikov product).
A double bond, however, can be protonated to generate two different cations, as shown in Figure 10-2. But which carbon in the double bond will receive the proton? (Or, alternatively phrased, which side of the double bond will become positively charged?)
The Russian chemist Vladimir Markovnikov observed that alkenes are protonated on the least substituted carbon in the double bond, generating the carbocation on the most highly substituted carbon atom. In other words, tertiary carbocations (carbocations substituted by three alkyl groups and abbreviated 3°) are preferred over secondary carbocations (cations substituted by two alkyl groups and abbreviated 2°). Secondary carbocations in turn are favored over primary carbocations (1°), those cations substituted by just one alkyl group.
Because of this preference for more highly substituted carbocations, halides add to the carbon that’s most substituted with alkyl groups. When addition occurs on the most substituted carbon, as in the reaction shown in Figure 10-2, the product is called the Markovnikov product, in honor of the discoverer of this phenomenon.
Because this reaction favors one of two products — halide addition to the more substituted side of the double bond as opposed to halide addition to the less substituted side — this reaction is said to be regioselective. Regioselective reactions are those that prefer one constitutional isomer in a reaction to another. (Recall that constitutional isomers are molecules with the same molecular formula, but the atoms bonded in different ways.)
But what accounts for this preference? Why are the more highly substituted carbocations more stable (and therefore preferable) to those cations with fewer alkyl substitutions? To understand this preference you need to look at the structure of the carbocation.
I’m Positive: Carbocations
Carbocations are an unstable species because they leave the cationic carbon two electrons short of filling the atom’s valence octet (and you know how much atoms hate that). In addition, the cationic carbon bears a full positive charge (which is something else that atoms don’t like). The carbocation carbon is, therefore, highly electron deficient.
Helping a neighbor: Hyperconjugation
Alkyl substituents, however, can donate some electrons to the carbocation, thereby stabilizing it. For carbocations substituted with alkyl substituents, instead of the molecule localizing the full positive charge on just the one carbon atom, some of the positive charge is shared by the neighboring alkyl substituents. This sharing of charge is energetically preferable to having all the charge localized on a single atom. Figure 10-3 shows the relative stability of primary, secondary, and tertiary carbocations.
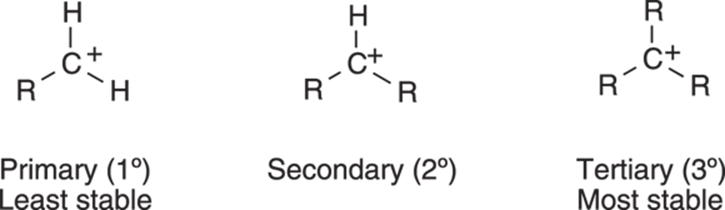
FIGURE 10-3: The relative stability of substituted carbocations.
Alkyl groups are able to donate electron density to the carbocation center through hyperconjugation. Hyperconjugation is organic-speak for the weak overlap that occurs between the empty p orbital on the carbocation and the σ bond of an adjacent alkyl C-H bond (or C-C bond), as shown in Figure 10-4. (Turn to Chapter 2 for a review of orbitals and bonds.) The more alkyl groups a carbocation center has, the more hyperconjugation takes place and the more the charge is shared; the more the charge is shared, the more stable the cation species is.
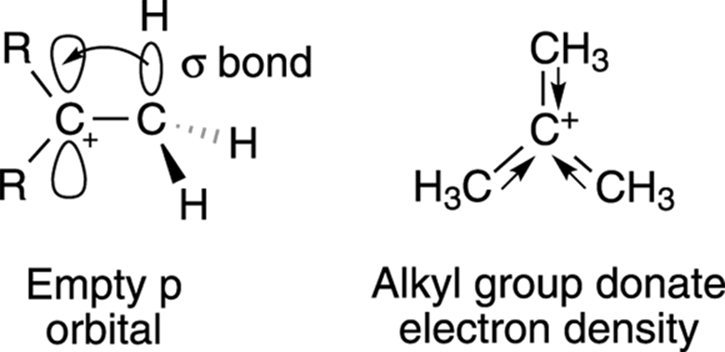
FIGURE 10-4: Hyperconjugation of neighboring alkyl groups.
Resonance stabilization of carbocations
Carbocations are also stabilized by resonance. (See Chapter 3 for how to draw resonance structures.) Carbocations with resonance structures are more stable than carbocations without resonance structures, all else being equal. Thus, benzylic cations (cations next to a benzene ring), andallylic cations (cations adjacent to a double bond) are stabilized cations because of resonance delocalization of the positive charge onto other atoms, as shown in Figure 10-5.
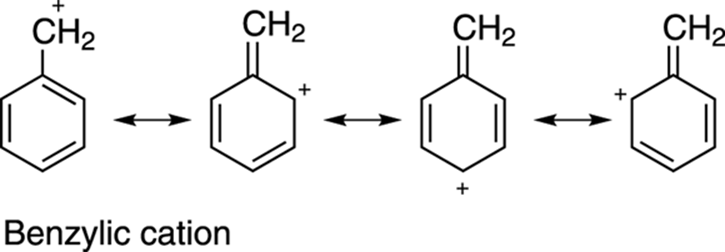
FIGURE 10-5: The resonance stabilization of the benzylic cation.
 Resonance structures stabilize molecules.
Resonance structures stabilize molecules.
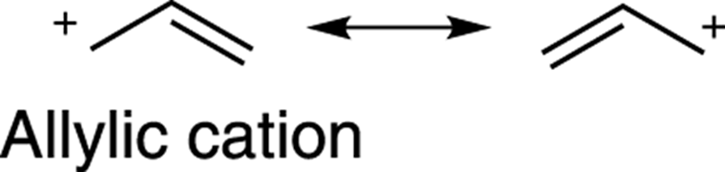
 The stabilities of carbocations can be approximated as follows (starting with the least stable and going to the most stable cation): primary cations < secondary cations ≈ allylic cations < tertiary cations ≈ benzylic cations.
The stabilities of carbocations can be approximated as follows (starting with the least stable and going to the most stable cation): primary cations < secondary cations ≈ allylic cations < tertiary cations ≈ benzylic cations.
Carbocation mischief: Rearrangements
Carbocations, like unsupervised children, have the capability of making a great deal of mischief. Take the reaction shown in Figure 10-6, for example. Reacting the alkene with hydrochloric acid yields the expected product only in small amounts; the major product is a rearranged alkyl halide.
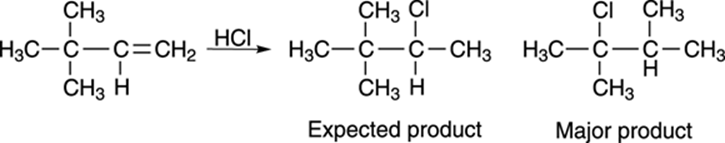
FIGURE 10-6: The addition of HCl to an alkene.
How did this reaction occur? Through a carbocation rearrangement. Alkyl substituents or hydrogens can (and will) shift from a neighboring carbon to the carbocation center whenever doing so generates a more stable carbocation. In this example, protonating the double bond as shown inFigure 10-7 makes the secondary carbocation (which is more stable than the primary cation, the alternative in this reaction). Shifting a methyl group (along with the two electrons in the bond) from the adjacent carbon to the secondary carbocation shifts the cation to the more stable tertiary carbon. The halide then attacks the tertiary cation to make the rearranged product.
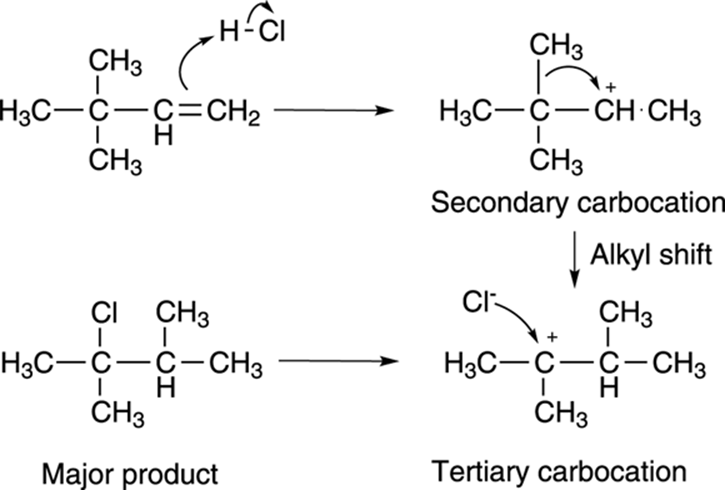
FIGURE 10-7: The mechanism of the carbocation rearrangement.
Spotting when these carbocation rearrangements may occur takes practice. When carbons adjacent to the cation center are highly substituted with alkyl groups, watch out for rearrangements.
Alkyl shifts can also open up small rings into larger ones, as shown in the reaction in Figure 10-8. Small rings (three- and four-membered rings) are less stable than medium-size rings (those containing five to six carbons) because of the ring strain from the constricted bond angles created by the small rings (recall that sp3 carbons prefer to have bonds at 109.5-degree angles to each other).
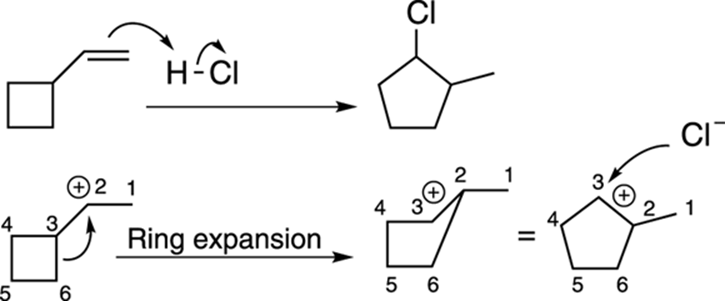
FIGURE 10-8: The carbocation rearrangement in a small ring.
Number all the atoms when working on ring expansion problems so you can keep track of all the atoms and keep the charges on the correct atoms in the rearranged product.
Alkyl rearrangements occur for two primary reasons:
· To make a more stable carbocation (for example, to change a secondary carbocation to a tertiary carbocation)
· To relieve ring strain (for cations next to small rings)
Adding Water across Double Bonds
Hydration, or adding water across a double bond to make an alcohol, is a reaction that’s similar to the addition of a hydrohalic acid across a double bond. Two different reactions accomplish the hydration. The first reaction adds the alcohol (OH group) to the most substituted carbon on the double bond to make the Markovnikov product, and the complementary reaction puts the alcohol on the least substituted carbon in the double bond to make the anti-Markovnikov product.
Markovnikov addition: Oxymercuration-demercuration
To make the Markovnikov product where the alcohol adds to the most substituted carbon, you react the alkene with mercuric acetate, Hg(OAc)2 and water, followed by addition of sodium borohydride, NaBH4 (see Figure 10-9).
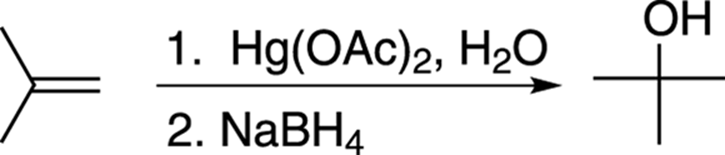
FIGURE 10-9: The oxymercuration-demercuration of an alkene.
The numbers over (or under) the reaction arrow indicate separate steps. In the case of oxymercuration-demercuration, the numbers specify that mercuric acetate is added first, followed by sodium borohydride. When no numbers are present over (or under) the arrow, this indicates that reagents are all added together in the same pot.
The mechanism for the oxymercuration-demercuration involves an attack of the double bond on the mercuric acetate to make a three-membered ring intermediate (called a mercurinium ion), as shown in Figure 10-10. Water then attacks the most highly substituted carbon to make the mercurial alcohol (after the loss of a proton). In the second step (when NaBH4 is added), sodium borohydride replaces the mercuric portion with hydrogen.
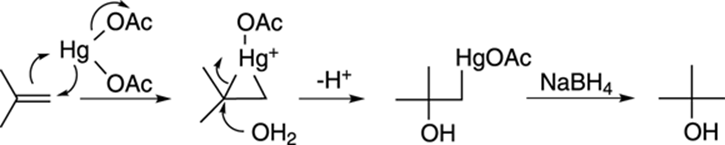
FIGURE 10-10: Oxymercuration-demercuration of an alkene.
Anti-Markovnikov addition: Hydroboration
With oxymercuration-demercuration, you have a reaction that converts alkenes into Markovnikov-product alcohols. To make the alcohol on the least-substituted carbon (called the anti-Markovnikov product) you use hydroboration, as shown in Figure 10-11. The addition of borane (BH3) in tetrahydrofuran solvent (THF) to the alkene, followed by the addition of hydrogen peroxide (H2O2) and sodium hydroxide (NaOH), make the anti-Markovnikov alcohol.

FIGURE 10-11: The hydroboration and oxidation of an alkene.
The mechanism for hydroboration involves the cyclic transition state shown in Figure 10-12. Borane adds to the least substituted side of the double bond to make the alkyl borane. Because the addition is concerted (both the hydrogen and BH2 are added simultaneously), the borane and hydrogen must add to the same face of the carbon-carbon bond (two groups adding to the same face of a double bond is called syn addition). In the second step, hydrogen peroxide (H2O2) in the presence of sodium hydroxide (NaOH) substitutes a hydroxyl group (OH) for the boryl unit (BH2) to make the anti-Markovnikov alcohol.
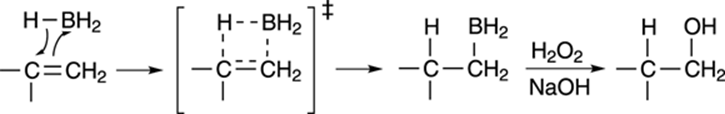
FIGURE 10-12: Mechanism of hydroboration and oxidation.
Syn addition is when two groups add to the same face of a double bond, and anti addition is where two groups add to opposite faces of a double bond. Therefore, syn addition to cycloalkenes results in the two added groups being cis to each other, and anti addition to cycloalkenes results in the two added groups being trans to each other.
A double shot: Dihydroxylation
You can add one hydroxyl group to a double bond using either oxymercuration-demercuration or hydroboration, but you can also add two hydroxy groups across a double bond using osmium tetroxide (OsO4) in the presence of hydrogen peroxide (H2O2), as shown in Figure 10-13. This reaction is called a dihydroxylation.
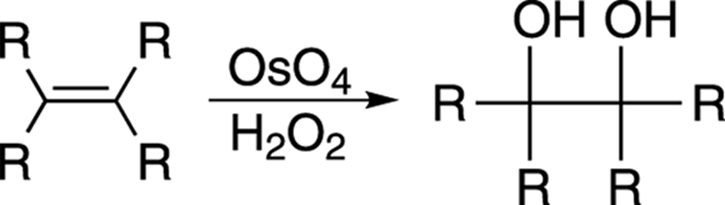
FIGURE 10-13: Dihydroxylation of an alkane.
The mechanism for dihydroxylation involves the reaction of the alkene and the osmium tetroxide to generate a five-membered cyclic osmate ester, as shown in Figure 10-14. Water regenerates the osmium tetroxide catalyst and makes the diol (a diol is a molecule with two hydroxyl [OH] groups). Because both oxygens in the osmium tetroxide are added to the same face of the double bond, both hydroxyl groups end up on the same side of the carbon-carbon bond (that is, they’re added via syn addition).
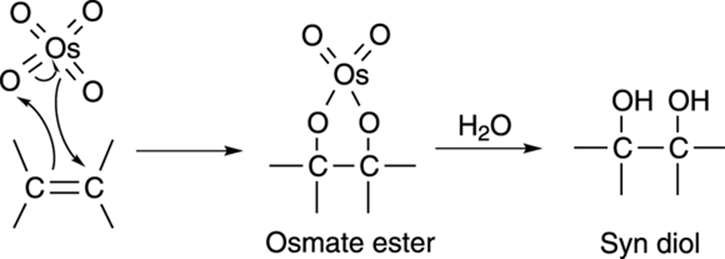
FIGURE 10-14: The mechanism for the hydroxylation of an alkene.
Double the fun: Bromination
Alkenes react rapidly with bromine (Br2) or chlorine (Cl2) in carbon tetrachloride (CCl4) solvent to make a dihalide, as shown in Figure 10-15. This reaction is, in fact, used as a test to see whether an unknown compound contains an alkene, because the blood-colored bromine (Br2) turns clear when it reacts with alkenes.
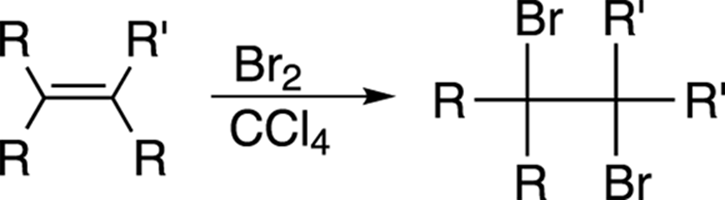
FIGURE 10-15: The bromination of an alkene.
The mechanism for bromination is somewhat unusual. Reaction of the alkene and bromine results in a cyclic three-membered intermediate called a bromonium ion (or chloronium ion for chlorine), plus the free halide, as shown in Figure 10-16. The free halide then attacks the backside of the bromonium ion to make the dibromide with anti-stereochemistry.
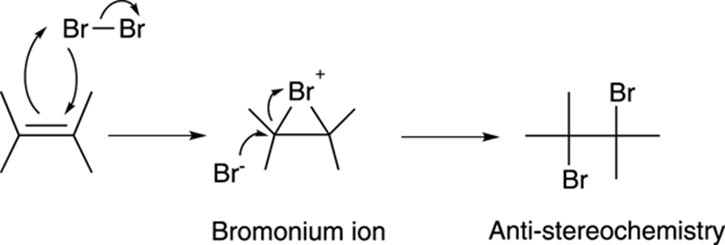
FIGURE 10-16: The mechanism for the bromination of an alkene.
Chopping Up Double Bonds: Ozonolysis
Ozonolysis is a way of cleaving carbon-carbon double bonds into two fragments using ozone (O3) as a reagent. The fragments formed are either aldehydes or ketones, depending on the nature of the R groups attached to the double bond (see Figure 10-17). If both R groups on one side of the double bond are alkyl groups, that side of the double bond will become a ketone fragment; if only one R group is an alkyl group and the other R is a hydrogen, that side of the double bond will become an aldehyde fragment.
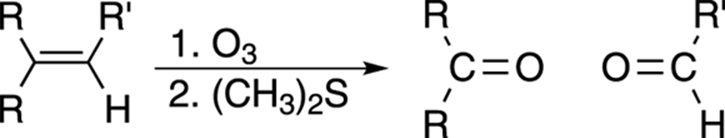
FIGURE 10-17: The ozonolysis of an alkene.
A quick way of determining the products of ozonolysis is to visually snip the double bond as shown in Figure 10-18, and then to cap both sides with oxygens to make the carbonyl compounds.
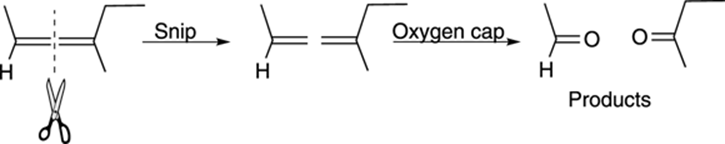
FIGURE 10-18: Determining products of ozonolysis.
A common exam problem is one that asks you to determine the structure of the starting alkene given the products of ozonolysis. If you remember how to determine the products of ozonolysis and then work backward from that reaction (snip off the oxygens, and then smoosh the two pieces together), you get the starting alkene.
Double-Bond Cleavage: Permanganate Oxidation
Permanganate oxidation, shown in Figure 10-19, performs essentially the same reaction as ozonolysis, except that permanganate is a somewhat stronger oxidizing reagent than ozone. In permanganate oxidation, all products that would have become aldehydes using ozonolysis are further oxidized to carboxylic acids with potassium permanganate (KMnO4); ketone fragments stay the same.
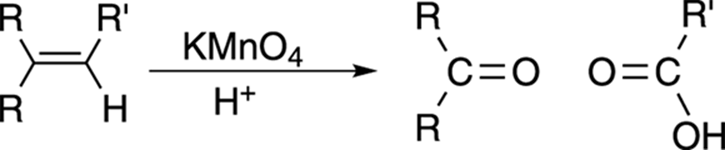
FIGURE 10-19: The permanganate oxidation of an alkene.
Making Cyclopropanes with Carbenes
Alkenes can be converted into cyclopropane rings by reacting with an unusual species called a carbene, as shown in Figure 10-20.
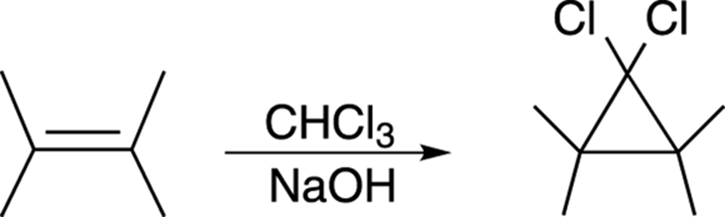
FIGURE 10-20: Making a cyclopropane ring from an alkene.
A carbene is a neutral carbon atom with two substituents and one lone pair of electrons. Dichlorocarbene (Cl2C:) is made by reacting chloroform (CHCl3) with a base like sodium hydroxide. The base deprotonates chloroform to make the conjugate base anion, which eliminates a chloride ion to make dichlorocarbene, as shown in Figure 10-21.
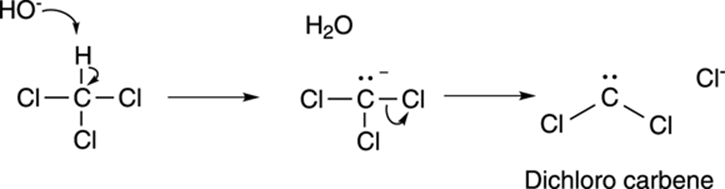
FIGURE 10-21: Making dichlorocarbene.
Dichlorocarbene can then react with double bonds to make cyclopropane rings, by the mechanism shown in Figure 10-22.
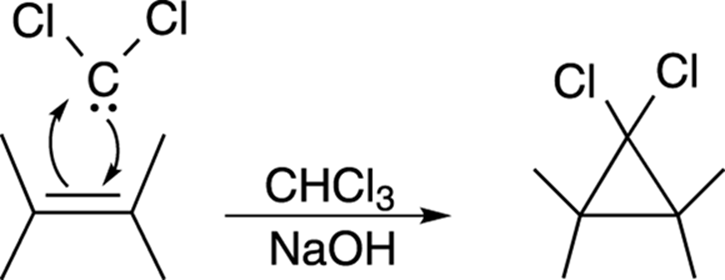
FIGURE 10-22: The dichlorocarbene addition to an alkene.
Making Cyclopropanes: The Simmons–Smith Reaction
A good way to make unsubstituted cyclopropanes from carbon-carbon double bonds is by using the Simmons–Smith reaction, shown in Figure 10-23. While the carbene methylene (H2C:) is not actually made in this reaction, a zinc species (ICH2ZnI) is used that reacts as if it were the carbene methylene. Because the reagent behaves like a carbene but really isn’t one, this zinc species is said to be carbenoid (carbene-like).
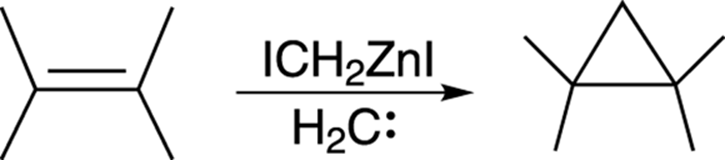
FIGURE 10-23: The Simmons–Smith reaction.
Making Epoxides
Epoxides are ethers in three-membered rings. They’re valuable synthetic reagents and are used in epoxy glues. Epoxides can be made by reacting an alkene with a peroxy acid (a carboxylic acid with an extra oxygen), as shown in Figure 10-24.
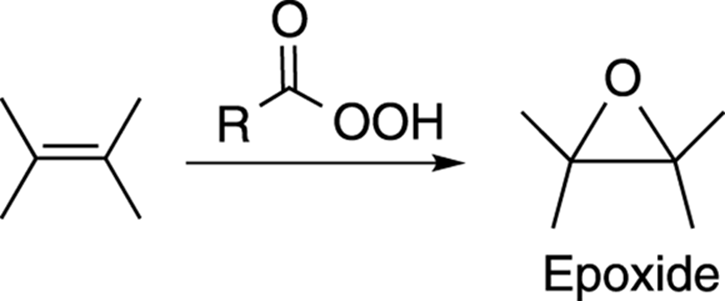
FIGURE 10-24: The epoxidation of an alkene.
Adding Hydrogen: Hydrogenation
Alkenes can be converted into alkanes by hydrogenation. Passing hydrogen gas through a solution containing a catalyst — usually palladium on carbon (Pd/C) or platinum (Pt) — and the alkene causes hydrogen to be added across the double bond in a syn addition (see Figure 10-25).
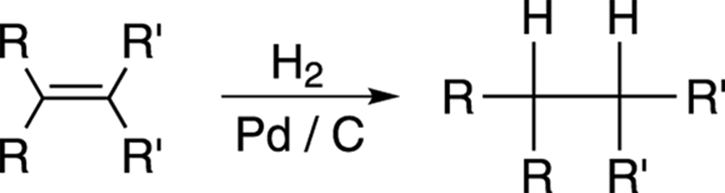
FIGURE 10-25: The hydrogenation of an alkene.