Organic Chemistry I For Dummies, 2nd Edition (2014)
Part III. Functional Groups
Chapter 15. Lord of the Rings: Aromatic Compounds
IN THIS CHAPTER
Seeing what makes aromatics so darn stable
Making molecular orbital diagrams for ring systems
Distinguishing aromatic, anti-aromatic, and non-aromatic rings
Naming aromatic compounds
According to organic legend, the structure of the parent aromatic system, benzene, came to the chemist August Kekulé in a dream. For a long time the structure of benzene — a liquid isolated from a tarry residue from burning petroleum gas — had eluded chemists, and many structures for the compound had been proposed by big names in the field. Kekulé claimed that in a dream he saw a snake devouring its own tail, forming a circle. Benzene must be a ring, he thought when he awoke, and jotted down the correct structure of the compound (or so he claimed, somewhat dubiously, many years later). Kekulé’s intuition about the cyclic structure of benzene was correct and soon led to the discovery of aromatic rings other than benzene.
Benzene and other aromatics are a highly interesting class of ringed compounds of exceptional stability. In this chapter, I show you how to name aromatics, and discuss aromatic compounds other than benzene that have properties and reactivities similar to those of benzene. I also show you how to determine whether a ring system is aromatic and show you what makes aromatic compounds so stable.
Defining Aromatic Compounds
Aromatics are a class of ring compounds containing double bonds. The name aromatic comes from the fact that many of the simple aromatic compounds that were first isolated were highly fragrant; the lovely odors of such substances as vanilla, almond, and wintergreen are due to the presence of aromatic compounds in these products. But many aromatic compounds are unpleasant smelling, or are odorless. Most simple aromatics, in fact, are obtained commercially from coal tar. Aromatic compounds contain double bonds, but they don’t react like alkenes, so they’re classified as a separate functional group. Benzene, for example, doesn’t behave as if it were 1,3,5-cyclohexatriene. Although bromine reacts rapidly with alkenes to make dibromides (see Figure 15-1), bromine is completely unreactive with benzene. This lack of reactivity results from aromatics being much more stable than alkenes.
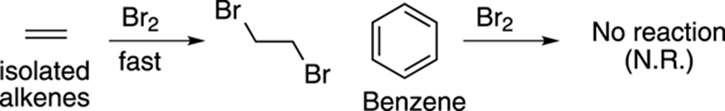
FIGURE 15-1: The relative stability of an alkene and benzene in the presence of bromine.
Because of this greater stability, aromatic compounds require significantly more vigorous reaction conditions to make them react compared to the conditions required to react simple alkenes. Aromatic rings scoff when exposed to the relatively mild reagents that react with alkenes (reagents such as those used in hydroboration, oxymercuration, and HBr addition, which are shown in Chapter 10). Trying to react aromatics with these reagents is like shooting popguns at a well-defended castle. These reagents simply glance off the rings and remain impotent in the reaction flask. You need to unleash the howitzer reagents to get aromatic rings to react, and I show you these in Chapter 16.
The structure of benzene
The actual structure of benzene is the hybrid of its two resonance structures, shown in Figure 15-2. (For a review of resonance structures, see Chapter 3.) Every bond in benzene is exactly the same length — neither as long as a single bond nor as short as a double bond — and its bond character can best be described as being 1.5, the bond length between that of a single bond and a double bond.
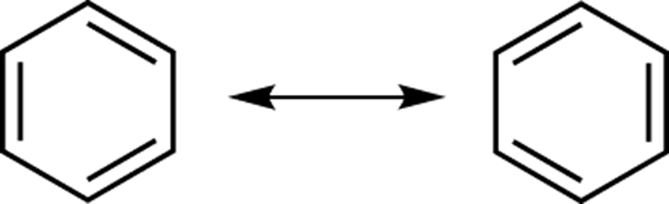
FIGURE 15-2: Benzene resonance structures.
Because benzene is a hybrid of two resonance structures, to more accurately show the location of the pi electrons with a single structure, all the double-bond electrons are often represented with a circle (as shown in Figure 15-3) rather than with individual double bonds. This circle drawing of benzene more accurately depicts the electron distribution of benzene because it shows all atoms and bonds as equal. But because the number of pi electrons is unclear in these drawings (and because reactions are better shown from the double bond–containing structures), I avoid using this circle representation for benzene in this book. Just keep in mind that when the double bond–containing structures for benzene are drawn, these hybrid structures are assumed.
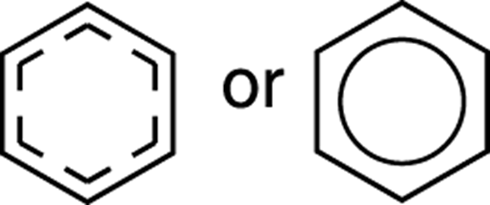
FIGURE 15-3: Benzene.
Diversity of aromatic compounds
Although benzene is the parent aromatic compound, many other aromatic compounds that have the same exceptional stability as benzene are known. Some of these aromatics consist of benzene rings smooshed together to make fused rings, such as benzopyrene (shown in Figure 15-4). These fused aromatic compounds, which are found in coal, have been found to be toxic. Chimney sweeps and other workers exposed to these compounds for long periods of time run the risk of developing cancers because of exposure to the benzopyrene and other aromatic compounds found in chimney soot.
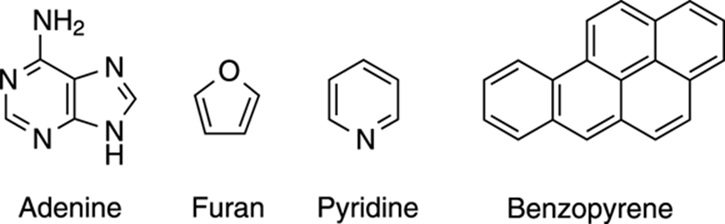
FIGURE 15-4: Some aromatic rings found in nature.
Aromatic compounds come in rings of all sizes, and many include heteroatoms (non-carbon atoms) like oxygen, nitrogen, and sulfur; aromatics that contain such heteroatoms include furan and pyridine, shown in Figure 15-4. All the DNA bases, including adenine, which is also shown inFigure 15-4, have aromatic character. Because they’re so stable, aromatic compounds are ubiquitous in nature.
So, what exactly makes a molecule aromatic?
At first, many chemists thought that all rings that contained alternating double bonds all the way around a ring would have the same aromatic character as benzene — and the same exceptional stability that comes along with this structure. This assumption is not correct, however. Cyclobutadiene, for example, shown in Figure 15-5, is an extremely unstable compound, and rapidly dimerizes (reacts with another molecule of cyclobutadiene) in solution via the Diels–Alder reaction from Chapter 14 (you get brownie points if you can draw the three-ringed product!). Benzene, as a result of its aromaticity, is more stable than its open chain analog, 1,3,5-hexatriene, but cyclobutadiene is actually less stable than its open-chain counterpart, 1,3-butadiene. Therefore, some rings are stabilized by these double bond–containing rings and become aromatic, while others (like cyclobutadiene) are destabilized and become what are called anti-aromatic rings.
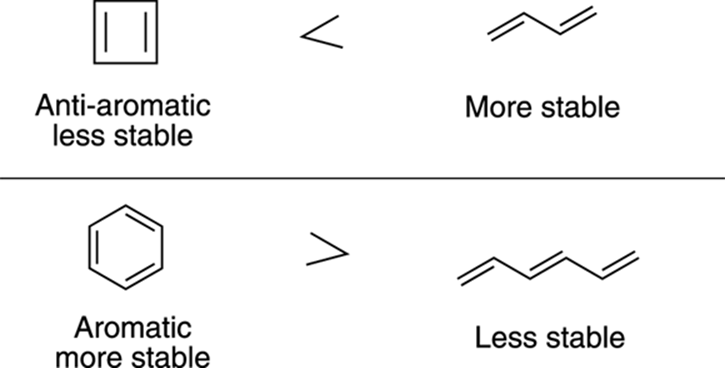
FIGURE 15-5: The relative stabilities of some ringed and open-chain alkenes.
Hückel’s 4n + 2 rule
A German chemist named Erich Hückel explained this somewhat confusing result. He observed that planar ring systems that contained 4n + 2 pi electrons (where n is an integer) were stabilized and aromatic, while ring systems with 4n pi electrons were destabilized and anti-aromatic. Thus, benzene, with its 6 pi electrons (2 pi electrons from each double bond), is aromatic because it follows the 4n + 2 rule, where n = 1. Cyclobutadiene, on the other hand, is anti-aromatic because of its 4 pi electrons; it follows the 4n equation, where n = 1.
Hückel’s equation, useful as it is, doesn’t explain why the rings that contain a Hückel number of pi electrons (4n + 2) have this stabilizing feature of aromaticity, while those with a non-Hückel number (4n) are destabilized. Although you know that benzene has resonance structures, and that resonance structures generally contribute to a molecule’s stability, the best explanation for the stability of aromatic compounds comes from molecular orbital theory, which is covered in the next section.
Explaining Aromaticity: Molecular Orbital Theory
Molecular orbital (MO) theory offers a more sophisticated model of bonding than the valence bond model (which is represented with Lewis structures). Of course, this greater sophistication comes at a cost: MO theory is not as user-friendly as the valence bond model. It’s been whispered (and not only among students) that while valence bond theory is too good to be true, MO theory is too true to be good. That description is somewhat unfair to both models, but it’s true that organic chemists have staved off using MO theory even when an MO model gives the correct description of a molecule and the valence bond model is wrong. For example, organic chemists prefer to use resonance structures to correct flaws in the valence model’s depiction of pi and nonbonding electrons rather than switch to the more precise (but more unwieldy) MO theory.
What the heck is molecular orbital theory?
In valence bond theory, electrons are located in bonds between atoms or as nonbonding electrons on an atom. In molecular orbital theory, electrons are not restricted to being localized between two atoms or on an atom itself. Instead, the electrons are allowed to spread across the entire molecule. If you think of orbitals as being like apartments for electrons, the electron has just been allowed to move from the small, cloistered hovel of valence bond theory into the much more spacious suite of molecular orbital theory. Because electrons can delocalize across the entire molecule in MO theory, the orbitals that these electrons reside in are called molecular orbitals, abbreviated Ψ (psi, pronounced “sigh,” as in, “Sigh! Why do I have to deal with molecular orbital theory?”).
 Valence bond theory and molecular orbital theory are just models for depicting the actual location of electrons in a molecule. Although valence bond theory is the more user friendly of the two, it’s not as accurate as MO theory because it doesn’t do as good a job at modeling how electrons can spread out over an entire molecule in certain instances (like in aromatic rings).
Valence bond theory and molecular orbital theory are just models for depicting the actual location of electrons in a molecule. Although valence bond theory is the more user friendly of the two, it’s not as accurate as MO theory because it doesn’t do as good a job at modeling how electrons can spread out over an entire molecule in certain instances (like in aromatic rings).
Making molecular orbital diagrams
A very informative orbital schematic is called a molecular orbital diagram. A molecular orbital diagram shows the number and relative energies of the different molecular orbitals for a molecule, and the electron occupancy in each of these orbitals. These diagrams are just like the atomic electron configurations you drew in introductory chemistry (reviewed in Chapter 2), except that these diagrams are for molecules instead of atoms, and in these diagrams you fill molecular orbitals rather than atomic orbitals.
To make an MO diagram, you need to follow three guidelines:
· Molecular orbitals are made by combining atomic orbitals (like s and p orbitals). The number of atomic orbitals combined must equal the number of molecular orbitals in the molecule. If you put six atomic orbitals in, for example, you must get six molecular orbitals out.
· The MOs that go down in energy are called bonding molecular orbitals; the MOs that go up in energy are called antibonding molecular orbitals.
· Electrons fill molecular orbitals in the same way that electrons fill atomic orbitals in an atom. In other words, the lowest-energy orbitals fill first with a maximum of two electrons per orbital, following Hund’s rule. (See Chapter 2 for a review of atomic orbitals and Hund’s rule.)
Every carbon atom in benzene is sp2 hybridized. Therefore, every carbon atom in benzene has one p orbital orthogonal (at a right angle) to the plane of the benzene ring, as shown in Figure 15-6. The pi orbitals in benzene are made from the overlap of these p orbitals. When making these MO diagrams for benzene and other aromatics, you usually only care about the pi orbitals (because only the pi electrons contribute to aromaticity). So, on these MO diagrams you leave out all the sigma molecular orbitals, and combine just the p orbitals to make your pi molecular orbitals (recall that only p orbitals make pi bonds).

FIGURE 15-6: Benzene p orbitals.
Two rings diverged in a wood: Frost circles
Because benzene has six p orbitals (one from each carbon), exactly six pi molecular orbitals must be formed from these six orbitals. A very handy guide to making the molecular orbital diagrams for these ring systems is a Frost circle. A Frost circle enables you to quickly place the six orbitals on the molecular orbital diagram and gives you the relative energies of the orbitals.
Making a Frost circle is easy: You simply draw a circle, and within the circle draw the carbon ring with one of its corners pointing down. Each point where the ring touches the circle is the location of one of the molecular orbitals. After you have all the orbitals situated at the correct energy level, you fill the orbitals with the number of pi electrons in the ring, and — voilà! — you have the molecular orbital diagram.
After drawing a couple of these molecular orbital diagrams for ring systems, you notice that in many diagrams two orbitals are at the same energy level. Such orbitals are called degenerate orbitals, using organic-speak.
Making the molecular orbital diagram of benzene
The construction of the molecular orbital diagram of benzene is shown in Figure 15-7. First, a circle is drawn, and then a six-membered ring is plopped inside the circle with the point down.
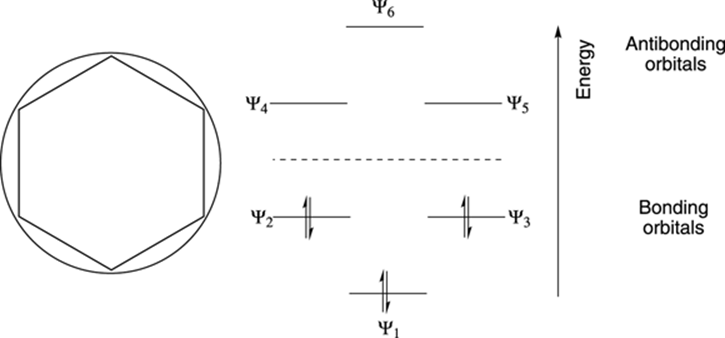
FIGURE 15-7: The Frost circle and the molecular orbital diagram of benzene.
 One of the most common mistakes in constructing MO diagrams using a Frost circle is to forget to put the point of the ring down in the circle.
One of the most common mistakes in constructing MO diagrams using a Frost circle is to forget to put the point of the ring down in the circle.
Every point where the six-membered ring touches the circle represents the location of one of the molecular orbitals (which I have transposed to the right of the circle in the figure for clarity). The orbitals lower than the center of the circle are called bonding orbitals, while those higher than the center of the circle are called antibonding orbitals. Bonding orbitals are lower in energy than antibonding orbitals. Because benzene has six pi electrons, you fill the lowest energy orbital (Ψ1) plus the two degenerate orbitals above it (Ψ2 and Ψ3). Note how lovely the electronic structure of benzene is — all three of these low-energy bonding orbitals are completely filled with electrons, while each of the high-energy antibonding orbitals remains empty. Because each of the filled orbitals in benzene is low in energy, benzene is exceptionally stable.
Seeing the molecular orbitals of benzene
Of course, these MO diagrams don’t tell you what the individual molecular orbitals look like. Each molecular orbital in benzene is made from a combination of the six atomic p orbitals. Recall that p orbitals have a node — a region of zero electron density at the center of the orbital where the wave function that describes the orbital changes sign. (This change in sign is made apparent by shading one half of the p orbital.) For a bonding interaction to occur between two p orbitals, the orbitals must line up so that each lobe that overlaps with another p orbital has the same sign. When orbitals line up with an opposite sign, it creates an antibonding (high-energy) interaction and a node between the orbitals. An example for the overlap of p orbitals is shown in Figure 15-8.
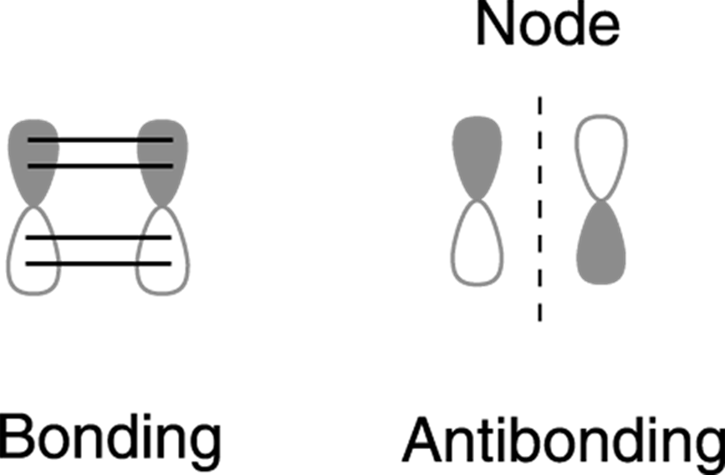
FIGURE 15-8: Bonding and antibonding p orbitals.
The MOs of benzene are shown in Figure 15-9. When all the p orbitals circle the benzene such that all the signs match up, every single orbital overlaps with the other in a bonding interaction and makes the molecular orbital (Ψ1) with the lowest possible energy. When one side of the ring has orbitals that line up one way, and the other side has orbitals that line up the opposite way, a molecular orbital is created that has one node across the benzene ring. The molecular orbitals Ψ2 and Ψ3 have one node each. A general feature of pi molecular orbitals is that the lowest energy level orbitals have zero nodes, the second energy level has one node, the third has two nodes, and so on. The more nodes a molecular orbital contains, the higher the energy of the orbital. In the highest energy molecular orbital of benzene (Ψ6), every p orbital is antibonding with its neighboring orbital.
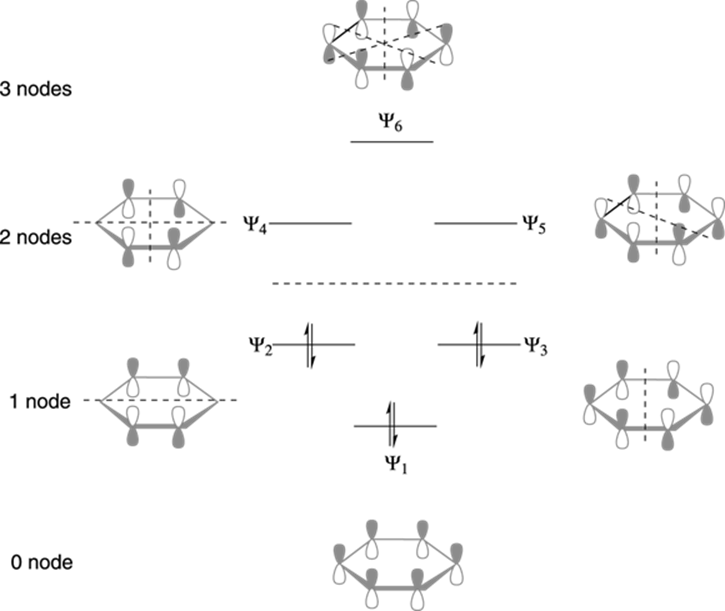
FIGURE 15-9: The molecular orbitals of benzene.
You might wonder how an electron is able to hop across nodes, those regions of zero electron density where electrons are not allowed. Continuing the apartment analogy, these nodes are like having rooms in your apartment, but no doorways. How do you get from one room into the next? The confusion comes from thinking of electrons only as particles, like little planets orbiting the nucleus sun, and ignoring the wave properties of electrons. Here a wave analogy is useful. When you thumb a guitar string on the fret of the guitar and then pluck the string, you see the vibration propagate past your thumb, a place where the vibration is zero. In the same way, an electron, because it can behave as a wave, can hop past the nodes in a molecular orbital. When thinking of node hopping, think of the electron as a wave rather than as a particle.
Making the molecular orbital diagram of cyclobutadiene
Now try making the MO diagram for cyclobutadiene, which is unstable and anti-aromatic (see Figure 15-10). You make the Frost circle the same as you did with benzene, except that you plunk a four-membered ring point down in the circle to obtain the MO levels. (Note that you get four molecular orbitals, because you start with four p atomic orbitals.) Because cyclobutadiene has four pi electrons (again, two from each double bond), you completely fill the first molecular orbital (Ψ1). The next two electrons are placed in the Ψ2 and Ψ3 orbitals, which are degenerate. According to Hund’s rule (refer to Chapter 2), you must place one electron into each degenerate orbital instead of pairing them in the same orbital. Thus, unlike benzene, in which all bonding orbitals are filled with electrons, two orbitals in cyclobutadiene are only partially filled. Cyclobutadiene, therefore, is essentially a diradical (a species containing two unpaired electrons) and diradicals are highly unstable! Certainly, you wouldn’t have guessed that from looking at the misleading Lewis structure.
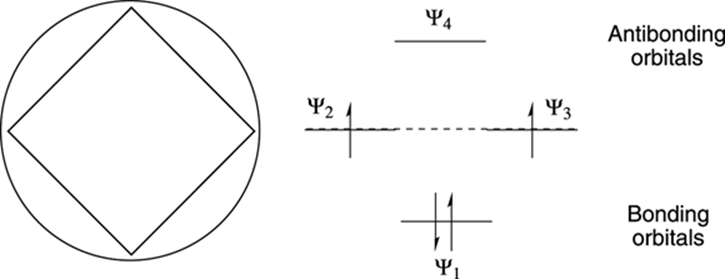
FIGURE 15-10: The Frost circle and the molecular orbital diagram of cyclobutadiene.
In fact, you see that this trend is followed for all aromatic and anti-aromatic rings. Aromatic rings have only filled bonding orbitals, while anti-aromatic rings have two unfilled orbitals, making them extremely grouchy and unstable.
Problem Solving: Determining Aromaticity
Now that I’ve shown you why aromatic rings are so stable, and why anti-aromatic rings are so unstable, I show how you can determine whether a ring system is aromatic, anti-aromatic, or non-aromatic.
For a molecule to be aromatic, it must meet the following four conditions:
· It must be a ring.
· It must be flat (planar).
· It must have in each atom of the ring a p orbital that’s orthogonal to the plane of the ring. In other words, no atom in the ring can be sp3 hybridized.
· It must have a Hückel number of pi electrons, following the 4n + 2 rule.
If the molecule meets the first three conditions, but only has 4n pi electrons, the molecule is anti-aromatic. If the molecule fails any or all of the first three conditions, then the molecule is non-aromatic. Following are some details about each of these points.
The first condition is that only rings can be aromatic. Acyclic systems cannot be aromatic.
The second condition involves the shape of the ring. Ringed systems can be flat or three-dimensional. Most conjugated ring systems are flat in order to maximize the overlap between the p orbitals. But some exceptions exist. Cyclodecapentaene, for example, shown in Figure 15-11, is puckered because two hydrogens in the ring are pushed into each other’s space, and the ring is forced out of planarity so that the hydrogens can occupy separate spaces. Because of its nonplanar structure, cyclodecapentaene is non-aromatic. Removing the two hydrogens and replacing them with a carbon bond to make naphthalene erases this problem; naphthalene is planar (and aromatic).
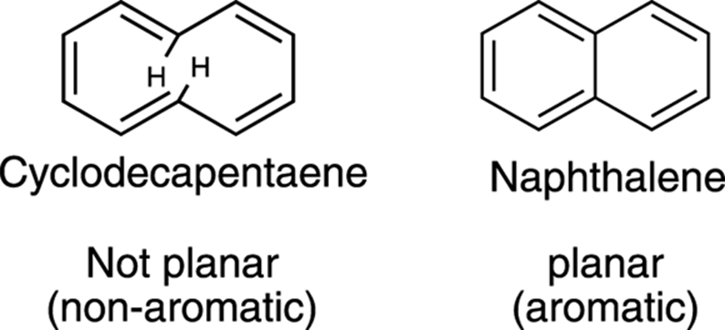
FIGURE 15-11: The nonplanar and planar rings of cyclodecapentaene and naphthalene.
Cyclooctatetraene (Figure 15-12) is another example of a molecule that has alternating double bonds around the ring but isn’t planar. Instead, this ring system adopts a puckered tub-shaped conformation. Why? Because if it remained flat, it would become anti-aromatic as a result of having a non-Hückel number of pi electrons (8)! Because anti-aromatic systems are so unstable, this compound puckers and sacrifices the p orbital overlap so that it won’t have to endure the instability associated with being anti-aromatic.
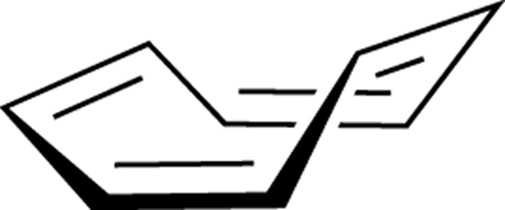
FIGURE 15-12: Cyclooctatetraene.
The third condition involves the orbitals. Aromatic systems must have an unbroken ring of p orbitals, so any ring that contains an sp3 hybridized carbon will not be aromatic. Cycloheptatriene, shown in Figure 15-13, is non-aromatic because one of the ring carbons is sp3 hybridized. However, carbocations (positively charged carbons) are sp2 hybridized (and have an empty p orbital), so the cycloheptatriene cation has an unbroken ring of p orbitals and is an aromatic compound.
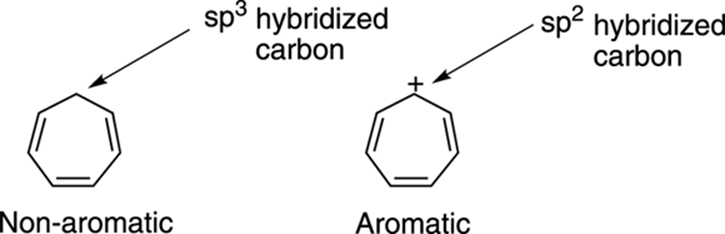
FIGURE 15-13: The nonaromatic cycloheptatriene molecule and the aromatic cycloheptatriene cation.
Finally, to be aromatic, a ring must have a Hückel number of electrons (4n + 2). Table 15-1 gives solutions to these equations for low values of n.
TABLE 15-1 Pi Electron Counts
|
Integer (n) |
Aromatic Numbers (4n + 2) |
Anti-aromatic Numbers (4n) |
|
0 |
2 |
— |
|
1 |
6 |
4 |
|
2 |
10 |
8 |
|
3 |
14 |
12 |
|
4 |
18 |
16 |
One tricky aspect of counting the number of electrons in the pi system is when the ring contains heteroatoms (like O, S, N). How do you know which lone pairs to count as part of the pi system and which to ignore (see Figure 15-14)?
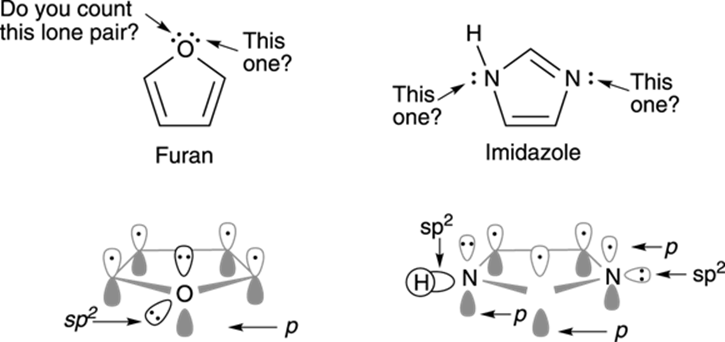
FIGURE 15-14: Counting pi electrons on heteroatoms.
The long answer is that you have to draw out the orbitals to see which lone pair electrons would go into the pi system and which ones wouldn’t. For furan, one lone pair from the oxygen would go into the p orbital (pi system), and one would go into an sp2 hybridized orbital orthogonal to the ring. So, the oxygen in furan would contribute two electrons to the pi system.
But wait a minute! Mustn’t the oxygen in furan be sp3 hybridized because the oxygen has four substituents (and lone pairs count as a substituent; see Chapter 2)? Not necessarily. Although the general rule of counting substituents to determine hybridization holds true in most cases, it fails when an atom with a lone pair is adjacent to a double bond (that is, when it’s conjugated). In those cases, the atom with the lone pair will rehybridize from sp3 to sp2 so that it can conjugate the lone pair of electrons with the double bond.
Both furan and imidazole have six pi electrons and are aromatic. (Can you look at the orbital diagrams in Figure 15-14 and see that that’s the case?)
 Here’s a timesaving tip so you don’t have to draw out the orbitals of heteroatom-containing rings every time. Heteroatoms (O, S, N) double bonded to other atoms in a ring cannot contribute their lone pairs to the pi system, because their p orbital is already being used to make the double bond. (Refer to Figure 15-14, and notice the orbitals of the double-bonded nitrogen in imidazole.) Heteroatoms only singly bonded to other atoms in a ring can contribute one lone pair to the pi system, but not two because only one lone pair can be in the pi system; the other lone pair will be in an sp2 hybridized orbital orthogonal to the pi system. (Refer to Figure 15-14, and notice the orbitals of the singly bonded oxygen in furan.)
Here’s a timesaving tip so you don’t have to draw out the orbitals of heteroatom-containing rings every time. Heteroatoms (O, S, N) double bonded to other atoms in a ring cannot contribute their lone pairs to the pi system, because their p orbital is already being used to make the double bond. (Refer to Figure 15-14, and notice the orbitals of the double-bonded nitrogen in imidazole.) Heteroatoms only singly bonded to other atoms in a ring can contribute one lone pair to the pi system, but not two because only one lone pair can be in the pi system; the other lone pair will be in an sp2 hybridized orbital orthogonal to the pi system. (Refer to Figure 15-14, and notice the orbitals of the singly bonded oxygen in furan.)
Problem Solving: Predicting Acidities and Basicities
Two of the most common problem types, when dealing with aromatic compounds, are predicting the acidities and the basicities of double bond–containing rings, including aromatic rings. These problems are designed to see if you can apply aromaticity on your own and understand that aromaticity is a stabilizing feature of molecules and ions.
Comparing acidities
One common question is to ask which one of two double bond–containing rings is more acidic, such as the molecules in Figure 15-15. As with any acid-base reaction in which you need to compare acidity, you should look to see which acid has the more stable conjugate base.
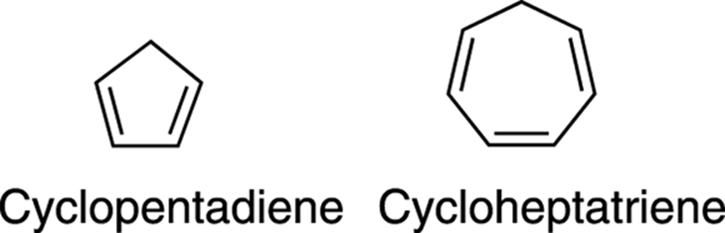
FIGURE 15-15: Cyclopentadiene and cyclohepta-triene.
Stronger acids have more stable conjugate bases.
Looking at the conjugate bases of both molecules (see Figure 15-16), you see that both compounds have rings that are entirely conjugated, but one has six pi electrons (cyclopentadienyl anion), while the other has eight pi electrons (cycloheptatrienyl anion). Thus, the conjugate base with six pi electrons is aromatic and should be more stable than the ring with eight pi electrons, which cannot be aromatic. Based on this analysis, cyclopentadiene must be more acidic than cycloheptatriene.
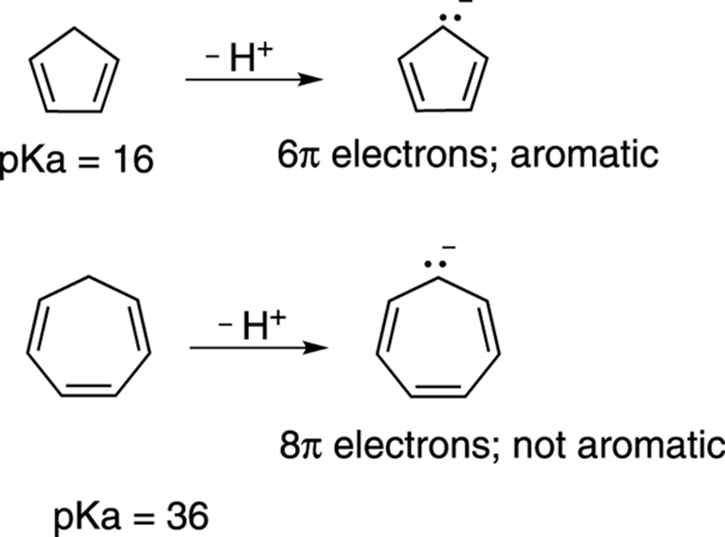
FIGURE 15-16: Comparing acidities of rings.
Comparing basicities
Another common question type is to compare or predict the basicity of nitrogen atoms in aromatic compounds. For example, can you predict which nitrogen is more basic in the aromatic molecule imidazole, shown in Figure 15-17?

FIGURE 15-17: Comparing the basicities of the nitrogens in imidazole.
To determine the strength of a base, you look at the stability of the conjugate acid. Protonating the bottom nitrogen in the imidazole in Figure 15-17 disrupts the aromaticity of the ring, because that nitrogen lone pair is part of the pi system. (Upon protonation, the lone pair is tied up in a bond to hydrogen and can no longer contribute to the aromaticity.) Protonating the top nitrogen doesn’t disrupt the pi system (or the aromaticity) of imidazole because the lone pair is situated in an sp2 hybridized orbital and is not part of the pi system. (Refer to Figure 15-14 for the orbital drawings of imidazole.) Therefore, the top nitrogen is much more basic than the bottom nitrogen, because the top nitrogen is much happier with an added hydrogen ion than the bottom nitrogen is. The general feature of these problems is that nitrogens whose lone pairs are not involved in the pi system are more basic than nitrogens with lone pairs in the pi system.
Naming Benzenes and Aromatics
Substituted benzenes are named with the parent name of — surprise! — benzene. Thus, benzene with an ethyl group is named ethylbenzene, as shown in Figure 15-18. When you have an alkyl chain that has more carbons than the aromatic ring, the alkyl portion becomes the parent group. When benzene is named as a substituent, it’s called a phenyl group (often abbreviated Ph), as in 3-phenylheptane. A general term for an aromatic ring as a substituent is the word aryl. A benzene with a methylene (CH2) portion is known as a benzyl group, as shown in Figure 15-19.
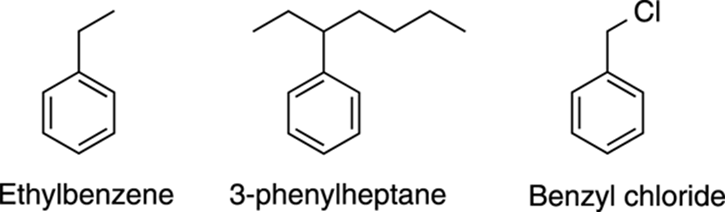
FIGURE 15-18: The names of some substituted benzenes.
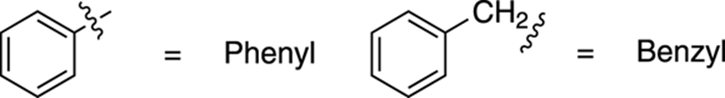
FIGURE 15-19: The phenyl ring and the benzyl group.
Don’t confuse a phenyl ring with a benzyl group.
Common names of substituted benzenes
One thing that you might find frustrating about aromatic compounds is that substituted benzenes are known by many common names. Methylbenzene, for example, is almost always called toluene. These names simply must be learned. Some of the most common substituted benzenes are shown in Figure 15-20.
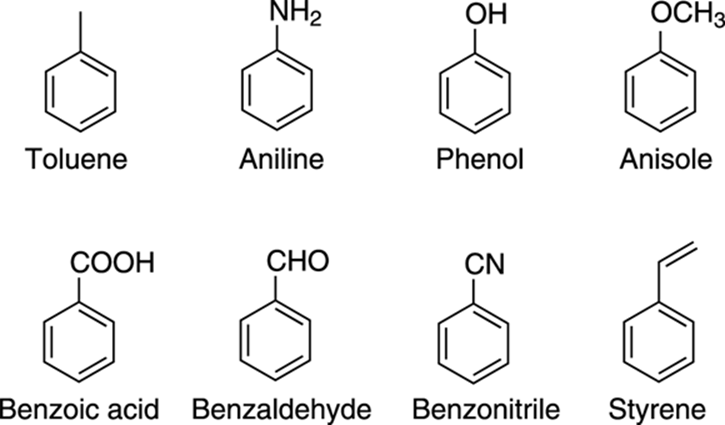
FIGURE 15-20: The common names of some substituted benzenes.
Many professors like to test whether you can remember the common names of compounds by giving synthesis problems on exams using the common names of the starting materials. For example, you might get a problem that says, “Starting with toluene, propose a synthesis to make compound X.” If you don’t know the structure of toluene, you’re up a creek on that problem without a you-know-what, even if you could have done the synthesis had you known the structure.
Names of common aromatics
The names of aromatics other than benzene are shown in Figure 15-21. For practice, you might want to see if you can determine how many pi electrons each of these rings contains. (They all have six pi electrons, but from where do these electrons come?)
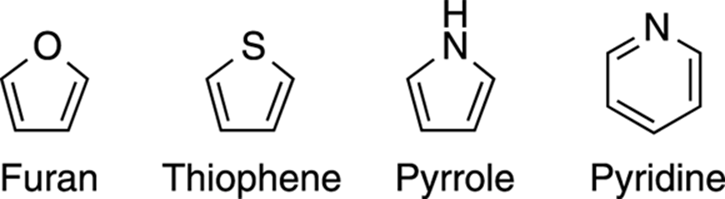
FIGURE 15-21: The names of some common aromatics.