Organic Chemistry I For Dummies, 2nd Edition (2014)
Part III. Functional Groups
Chapter 16. Bringing Out the Howitzers: Reactions of Aromatic Compounds
IN THIS CHAPTER
Seeing electrophilic aromatic substitution reactions of aromatics
Working through the synthesis of polysubstituted benzenes
Seeing nucleophilic aromatic substitution reactions
In Chapter 15, I discuss how you can determine whether a ring system is aromatic and what makes such aromatic compounds so stable. In this chapter, I show you how you can get such aromatic compounds to react by introducing a much hotter set of reagents than those used to react normal alkenes. These aromatic reactions are used to introduce multistep synthesis problems, or problems that require you to synthesize a complex structure by stringing together a sequence of known reactions.
Electrophilic Aromatic Substitution of Benzene
Unlike the double bonds of alkenes, the double bonds in benzene are only weakly nucleophilic (nucleus loving), so you need powerful electrophiles (electron lovers) to make benzene react. You usually have to throw a full positive charge on the electrophile to make it strong enough to react with benzene. For example, Br2 reacts with alkenes but does not react with benzene. However, positively charged bromine, Br+, does react with benzene, because positively charged species are much more electrophilic than neutral species are. Unlike the reaction of bromine with alkenes (in which you get an addition reaction across the double bond), when benzene is reacted with the bromine ion (Br+), you get substitution reactions (in which the electrophile substitutes for hydrogen).
Reacting benzene with electrophiles is a reaction called electrophilic aromatic substitution, and the general mechanism for this reaction is shown in Figure 16-1. In the first step of the mechanism, one of the double bonds in benzene attacks the positively charged electrophile (E+) to make a cation. This intermediate cation is stabilized by resonance (can you draw out the other two resonance structures?), but it’s non-aromatic (because the ring has an sp3-hybridized carbon). In the second step, when a base (abbreviated B: –) plucks off a proton adjacent to the carbocation, the aromatic ring is re-formed to make the substituted benzene.
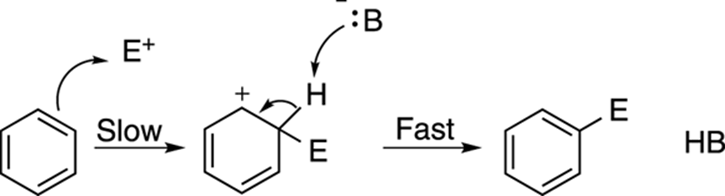
FIGURE 16-1: The mechanism of electrophilic aromatic substitution.
Figure 16-2 shows the reagents that you use to make different types of positively charged electrophiles. These reagents can be used to nitrate, brominate, chlorinate, or sulfonate benzene rings. These electrophiles all add to benzene in the fashion shown in Figure 16-1. The alkylation and acylation reactions are discussed in more detail in the following sections.
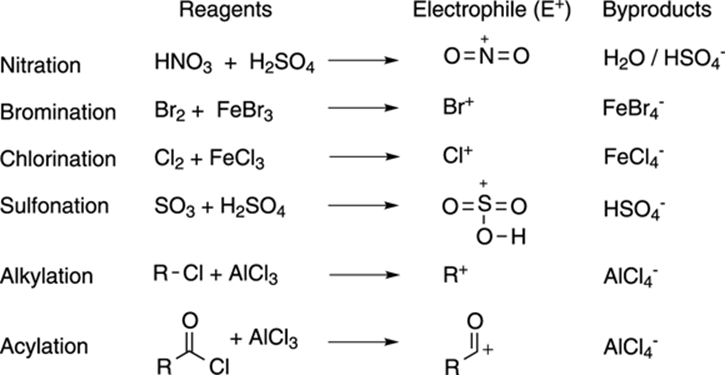
FIGURE 16-2: Generating electrophiles for electrophilic aromatic substitution reactions.
Adding alkyl substituents: Friedel–Crafts alkylation
One way to alkylate benzene rings was proposed by Charles Friedel and James Crafts. Reacting alkyl chlorides with the Lewis acid aluminum trichloride makes carbocations, they discovered, as shown in Figure 16-3. Carbocations are electrophilic enough to react with benzene to form alkyl benzenes by the same mechanism outlined in Figure 16-1.
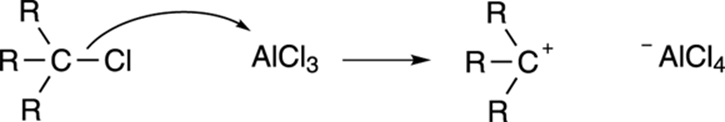
FIGURE 16-3: Making carbocations.
Carbocations, though, are like naughty little children. As illustrated in Chapter 10 with the addition of HCl to alkenes, carbocations can — and will — rearrange if doing so makes a more stable carbocation.
 Tertiary cations are more stable than secondary cations, which are more stable than primary cations.
Tertiary cations are more stable than secondary cations, which are more stable than primary cations.
For example, reacting benzene with propyl chloride and aluminum trichloride makes isopropyl benzene as the major product, with the expected product, propyl benzene, as the minor product. See Figure 16-4 for the products of this reaction.
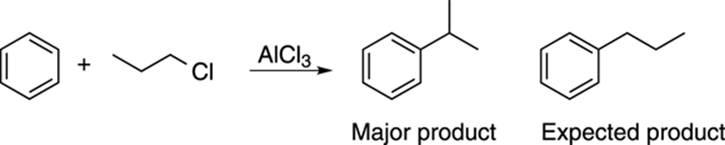
FIGURE 16-4: The Friedel–Crafts alkylation.
The reason for this reaction not giving the expected product is that the primary cation can rearrange to the more stable secondary carbocation by a hydride shift, as shown in Figure 16-5. The addition of this secondary cation to benzene yields isopropyl benzene as the major product. Because of these rearrangements, adding straight-chain alkyl groups to benzene is difficult using the Friedel–Crafts alkylation reaction.
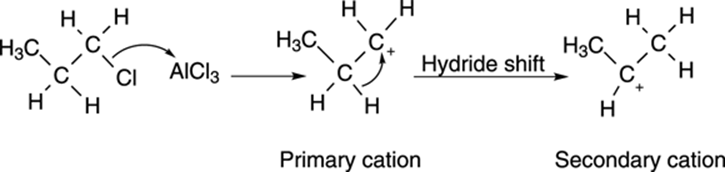
FIGURE 16-5: The carbocation rearrangement in the Friedel–Crafts reaction.
Overcoming adversity: Friedel–Crafts acylation
To stop these pesky rearrangements that lead to undesired products, Friedel and Crafts came up with an acylation reaction (pronounced ay-sill-ay-shun). Taking an acid chloride (RCOCl) and adding aluminum trichloride makes an acylium cation, as shown in Figure 16-6. Acylium ions are stabilized by resonance and don’t rearrange.
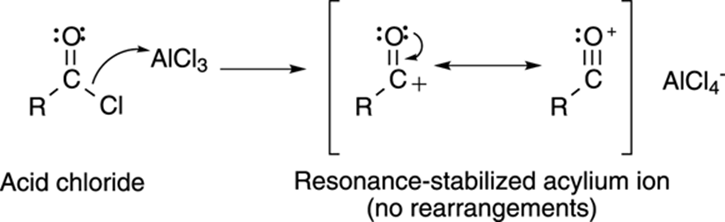
FIGURE 16-6: Making acylium cations.
These acylium ions then react with benzene to make aryl ketones, as shown in Figure 16-7. Aryl ketones can be conveniently reduced with hydrogen and palladium on carbon (Pd/C) to alkyl aromatics. (Regular ketones not next to benzene rings, however, are untouched by these reagents.) Although it requires an additional step, this reaction sequence — acylation followed by reduction — is a handy way of making alkyl benzenes without worrying about carbocation rearrangements giving you undesired products.
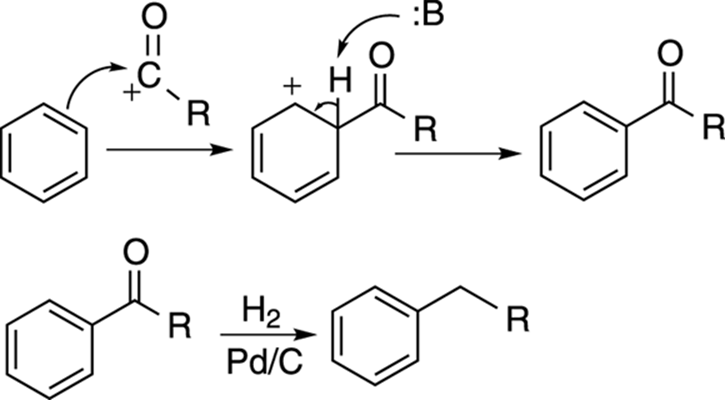
FIGURE 16-7: The Friedel–Crafts acylation mechanism and a convenient follow-up reaction to reduce the aryl ketone to the alkane.
Reducing nitro groups
A convenient way of making aryl amines is by reducing nitro groups. These groups can easily be reduced by the addition of tin chloride (SnCl2) in a little acid, as shown in Figure 16-8.
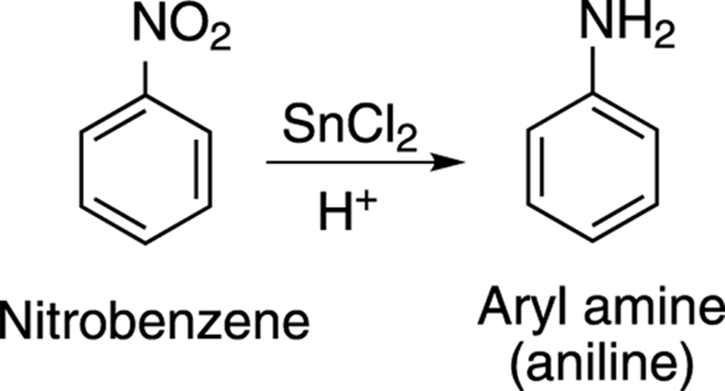
FIGURE 16-8: Nitro reduction and the formation of an aryl amine.
Oxidation of alkylated benzenes
Potassium permanganate is a powerful oxidizing reagent. In the presence of acid, this reagent takes alkyl benzenes, chews them up, and spits out aryl carboxylic acids (benzoic acids; refer to Figure 15-20), as shown in Figure 16-9. Any alkyl side chain that has a hydrogen adjacent to the phenyl ring will be oxidized to a carboxylic acid (a COOH group). If the alkyl side chain has no hydrogen adjacent to the ring, it will be left untouched.
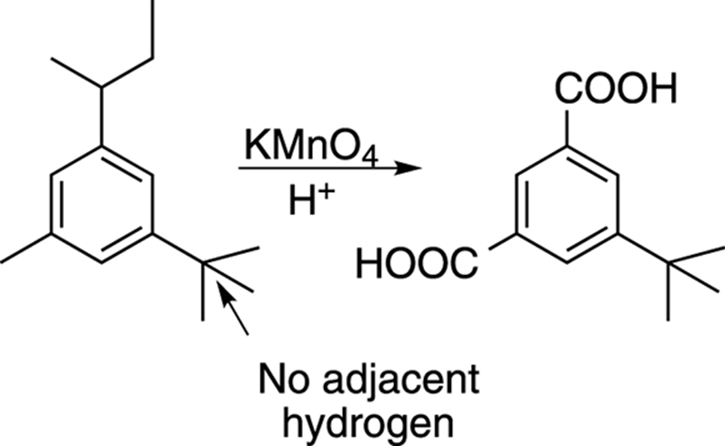
FIGURE 16-9: Permanganate oxidation of alkyl benzenes.
Adding Two: Synthesis of Disubstituted Benzenes
Now you know the reagents that add different groups to benzene. But what if you want to synthesize disubstituted benzenes? After adding the first group, three locations are possible for the next group to add to, as shown in Figure 16-10. One possibility is for the next group to add ortho— organic-speak for adding to the carbon adjacent to the substituent. The group could also add meta (two carbons away from the first group) or it could add para (on the opposite side of the ring from the original group).
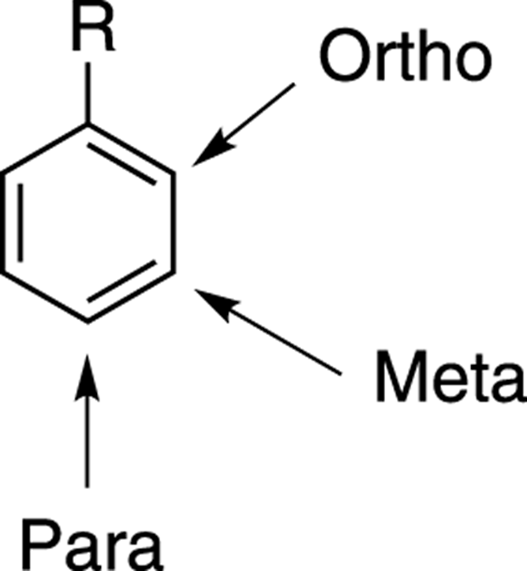
FIGURE 16-10: The ortho, meta, and para positions.
Where the second substituent adds — ortho, meta, or para to the first substituent — depends on the nature of the group already attached to the benzene ring. Think of the substituents already on the ring as traffic controllers at an airport, directing the electrophile to land at certain runways (ortho, meta, or para, in this case). Electron-donating substituents activate the ring (that is, they make the ring react faster than an unsubstituted benzene reacts), and they tell the incoming electrophile to land either at the ortho or the para position. With electron-donating substituents, you usually get a mixture of the ortho and para products. Electron-withdrawing groups on the ring, on the other hand, deactivate the benzene ring toward electrophilic attack (making the reaction slower than is the case with an unsubstituted benzene) and these electron-withdrawing groups tell the incoming electrophile to land at the meta position.
Why do you get different substitution products depending on whether the ring is substituted with electron donors or electron acceptors? The answer is that electrophiles add to the benzene ring on the atom that generates the most stable intermediate cation. With electron donors on the ring, the intermediate carbocation resulting from ortho or para addition is more stable than the cation deriving from meta addition. With an electron-withdrawing substituent on the ring, the intermediate carbocation from meta addition is more stable than the carbocation resulting from ortho or para addition.
In the following sections, I show you why the cation resulting from ortho-para addition with electron donors is more stable than the meta-derived cation. I also discuss why the cation resulting from meta addition with electron acceptors is more stable than the cations resulting from ortho or para addition.
Electron donors: Ortho-para activators
If you brominate anisole, for example, as shown in Figure 16-11, you get substitution of the bromine at the ortho and para positions, but not to the meta position. This is because methoxy groups (OCH3) are pi electron donors, so they direct all incoming electrophile traffic into the ortho and para positions.
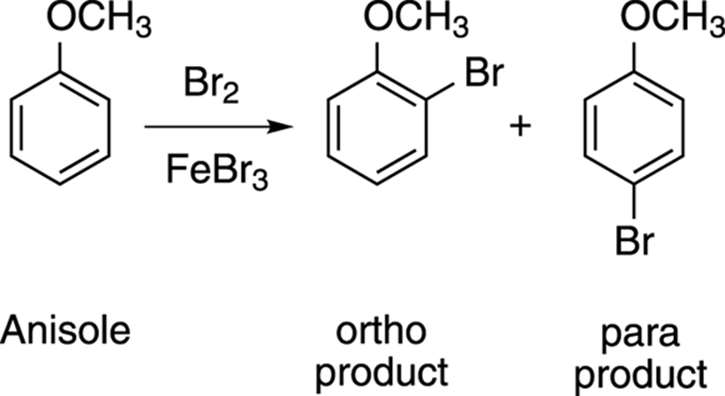
FIGURE 16-11: Bromine addition to anisole.
You can see why the methoxy group directs to the ortho and para positions by looking at the intermediate carbocation for both the para substitution and the meta addition (see Figure 16-12). With para substitution (and with ortho substitution), a much more stable intermediate carbocation is formed compared to the cation that’s formed when the substituent adds in the meta position. The intermediate carbocation that results from para substitution has four resonance structures, shown in Figure 16-12, with one of these resonance structures being particularly good because all valence octets on all atoms are filled. (This good resonance structure is boxed in the figure.) The carbocation resulting from meta substitution, on the other hand, has only three resonance structures, none of which have all atoms with filled valence octets. Recall the general rule that stability increases as the number of resonance structures increases. Thus, with electron donors on the aromatic ring, ortho-para products are selectively formed.
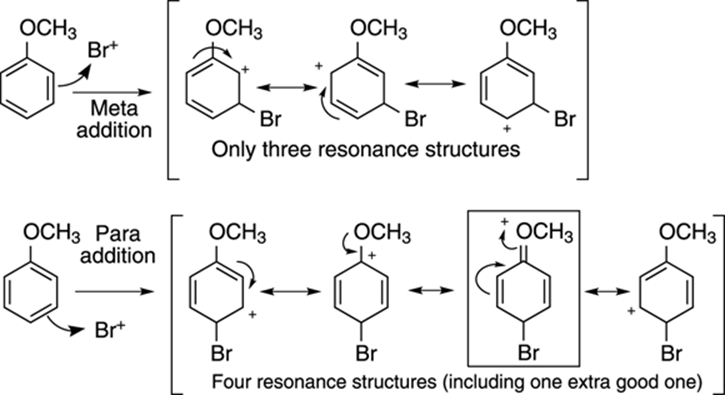
FIGURE 16-12: The relative stabilities of intermediate carbocations resulting from meta and para substitution of anisole.
Electron-withdrawing groups: Meta directors
Of course, the situation is reversed when the aromatic ring is substituted with electron-withdrawing substituents rather than electron-donating substituents. Electron-withdrawing substituents usually direct incoming electrophiles to the meta position. Take the bromination reaction of nitrobenzene, for example, shown in Figure 16-13. Nitro groups are electron-withdrawing groups, so bromine adds to the meta position.
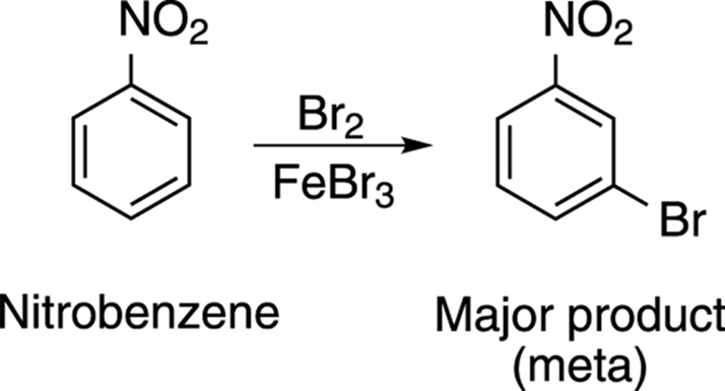
FIGURE 16-13: The bromine substitution of nitrobenzene.
To see why the meta product is formed instead of the ortho-para products, compare the intermediate cation formed as a result of para bromine addition to the cation generated from meta addition (see Figure 16-14). The intermediate carbocation resulting from para substitution (or ortho substitution) has three resonance structures, but one of them is a particularly bad resonance structure because the structure has two adjacent positive charges (and like charges repel). Thus, this bad resonance structure doesn’t contribute much to the overall resonance hybrid. The cation resulting from meta substitution also has three resonance structures, but none of them is bad. Thus, for benzenes substituted with electron-withdrawing groups, the cation resulting from meta substitution is more stable than the cation resulting from either ortho or para substitution.
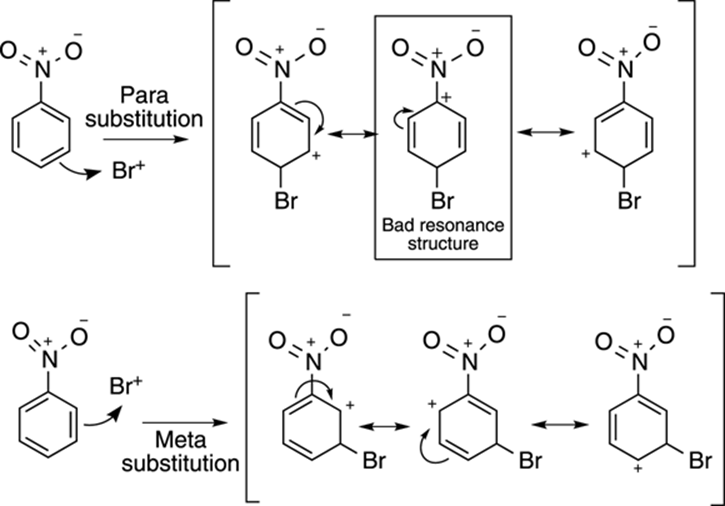
FIGURE 16-14: The relative stabilities of intermediate carbocations resulting from para and meta substitution to nitrobenzene.
The main point to remember here is that electron-donating groups direct substitution to the ortho and para position, while pi electron-withdrawing groups direct substitution to the meta position.
So far, I’ve talked about electron-donating and electron-withdrawing substituents, without really saying what I mean. Any substituent whose first atom (the one that’s attached to the benzene ring) has a lone pair will be a pi electron donor to the phenyl ring, as shown in the resonance structure in Figure 16-15. Note that the resonance structures show that substituents that are pi donors add electron density to the ortho and para positions of the ring. Thus, pi donors activate the benzene ring toward electrophilic attack (attack by incoming electrophiles) at the ortho and para positions. Adding electron density to the benzene ring makes the ring more nucleophilic (that is, more nucleus loving) and activates the ring. Therefore, pi donors are considered ring activators.
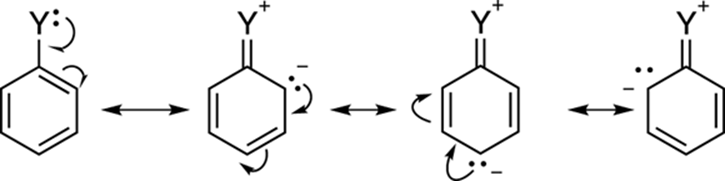
FIGURE 16-15: Pi electron donors to the phenyl ring.
The only exceptions are the halogens, which are not terribly good pi donors. They deactivate the ring as a result of being highly electronegative groups, pulling electrons away from the benzene ring toward themselves, making the ring less nucleophilic. But even though halogens are ring deactivators, they’re still ortho-para directors.
Pi-withdrawing groups (such as NO2 groups, carbonyl groups, CN, and so on) pull electrons away from the ring and deactivate it, making the ring less nucleophilic. Pi electron-withdrawers are thus ring deactivators. A deactivator means that the reaction of benzenes substituted with these substituents will be slower than the reaction of unsubstituted benzene. Pi electron-withdrawing substituents are meta directors. Table 16-1 outlines the nature of different substituents.
TABLE 16-1 The Nature of Aromatic Substituents
|
Ortho-Para Directing |
Meta Directing |
||
|
Strongly activating |
Weakly activating |
Deactivating |
Deactivating |
|
OH |
Alkyl |
Halogens (F, Cl, Br, I) |
NO2 |
|
OCH3 |
Phenyl |
—COR (COOH, COOR, CHO, and so on) |
|
|
NH2 |
CN |
||
|
NR2 |
SO3H |
||
 Here’s a tip for remembering Table 16-1: Any substituent whose first atom (the one attached to the benzene) contains a lone pair of electrons will be an ortho-para director (although not necessarily a ring activator). Those substituents without a lone pair on the first atom will likely be meta directors (with the exception of alkyl groups and aromatic rings, which are ortho-para directors).
Here’s a tip for remembering Table 16-1: Any substituent whose first atom (the one attached to the benzene) contains a lone pair of electrons will be an ortho-para director (although not necessarily a ring activator). Those substituents without a lone pair on the first atom will likely be meta directors (with the exception of alkyl groups and aromatic rings, which are ortho-para directors).
For performing multistep synthesis, the reduction of a nitro group to make an aryl amine is a way of converting a meta director into an ortho-para activator. The oxidation of an alkylated benzene (to make an aryl carboxylic acid) is a way of converting an ortho-para activator into a meta director. Additionally, the reduction of an aryl ketone (the product of a Friedel–Crafts acylation) to an alkane is a way of converting a meta director into an ortho-para activator.
Problem Solving: Synthesis of Substituted Benzenes
When thinking about the synthesis of polysubstituted benzenes (benzenes substituted more than once), remember that the order of substituent addition is crucial. You have to put on the substituents in the right order so that they direct the next substituent to the right place.
For example, how would you synthesize 3-bromo-1-ethylbenzene, shown in Figure 16-16? First, you should notice that the substituents are meta to each other. That means that the first substituent that you add should be a meta director in order to get the next substituent in the meta position. But the substituents — the ethyl group and the bromine — are both ortho-para directors! How are you going to get them meta to each other? At some point, one of them must have been a meta director that then got converted into the ortho-para substituent.
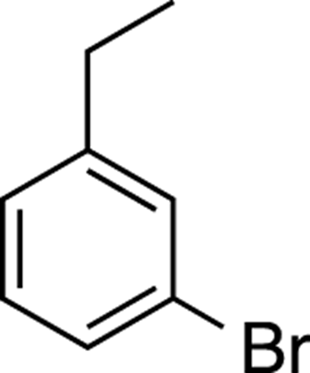
FIGURE 16-16: 3-bromo-1-ethylbenzene.
In the case of 3-bromo-1-ethylbenzene, the ethyl group could be added by a Friedel–Crafts acylation, as shown in Figure 16-17. Acyl groups are meta directors. The bromine could then be added and would go meta to the acyl group. The acyl group could then be reduced into the alkyl portion, as shown in the last step of Figure 16-17. The key to this synthesis is the order — switching any of the steps would lead to the wrong product.
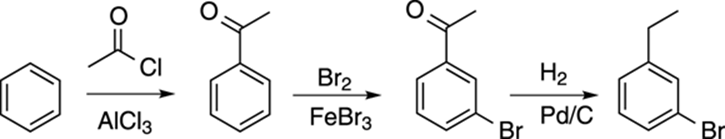
FIGURE 16-17: The synthesis of a disubstituted benzene.
The key to mastering aromatic synthesis problems is to add substituents in the correct order.
Nucleophiles Attack! Nucleophilic Aromatic Substitution
I’ve shown you how benzene can act as a nucleophile (nucleus lover), reacting with powerful electrophiles in the electrophilic aromatic substitution reaction. Benzene can also act as an electrophile when the ring is sufficiently activated toward nucleophilic attack. When benzene is activated toward nucleophilic attack, strong nucleophiles can displace leaving groups substituted on the ring (usually a halide).
Benzene rings activated toward nucleophilic attack include those that are substituted with strong electron-withdrawing groups in the ortho and para positions (groups like NO2, CN, COR, and so on). Substituted benzenes that are not activated with powerful electron-withdrawing groups will not undergo nucleophilic aromatic substitution. For example, 1-bromo-2,4-dinitrobenzene, shown in Figure 16-18, is activated toward nucleophilic attack because it has two powerful electron-withdrawing groups (NO2) ortho and para to the leaving group (Br). The addition of hydroxide ion (HO–) displaces the bromide.
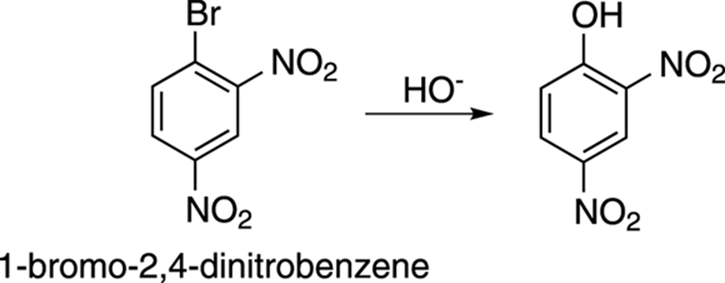
FIGURE 16-18: Nucleophilic aromatic substitution.
The mechanism of the addition is shown in Figure 16-19. First, the nucleophile (HO–) attacks the benzene ring at the carbon holding the leaving group, generating a carbanion (a negatively charged carbon). This carbanion is stabilized by the electron-withdrawing groups (NO2) on the ring (and by additional resonance structures that are not shown). The anion can then displace the bromide ion leaving group to regenerate the substituted aromatic ring.
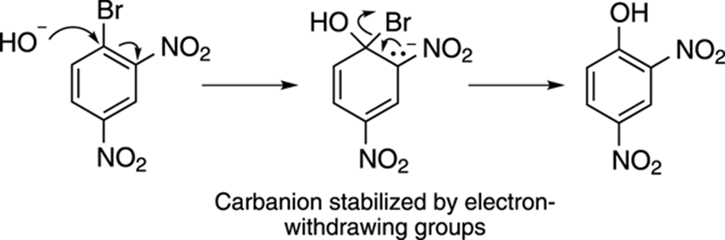
FIGURE 16-19: The mechanism of nucleophilic aromatic substitution.
Nucleophilic aromatic substitution only occurs with benzene rings activated by strong electron-withdrawing groups.
A somewhat less useful but still highly interesting reaction of benzene is the reaction of a halobenzene (like bromobenzene or chlorobenzene) with a strong base (like hydroxide) at high temperature and pressure. Because the aromatic ring is not activated for nucleophilic attack by electron-withdrawing substituents, you can’t get nucleophilic aromatic substitution. Instead, you get elimination, as shown in Figure 16-20, to make a weird intermediate called a benzyne. Benzyne is a highly unstable intermediate, consisting of a benzene ring with a triple bond. Nucleophiles (abbreviated –:Nuc in Figure 16-20) quickly add to benzyne to make substituted benzenes.
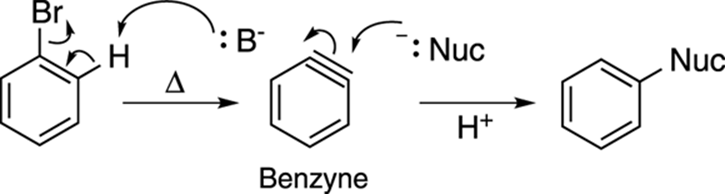
FIGURE 16-20: Benzyne reactions.
An example of benzyne addition is shown in Figure 16-21. In this case, the base that’s used to make the benzyne and the nucleophile that reacts with the benzyne is hydroxide (–OH). Note that because the nucleophile can add to either side of the benzyne, you get a mixture of products. Because you often get product mixtures with substituted benzenes, and because this reaction requires high temperature and pressure, this reaction is not quite as generally useful as electrophilic or nucleophilic aromatic substitution.
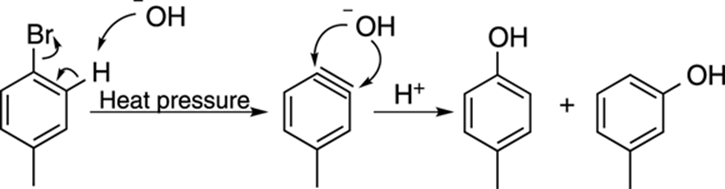
FIGURE 16-21: An example of benzyne substitution.