Physical Chemistry: A Very Short Introduction (2014)
Chapter 4. States of matter
A physical chemist is interested in all the ‘states’ of matter, the physical form in which it is found. There are three traditional states: gas, liquid, and solid. There are well-known borderline states, such as the liquid crystals that are used on many visual displays, which are half-liquid and half-solid. A new arrival in the solid category consists of nanomaterials, matter present on a scale of about 100 nm and less. Another new state is almost pure surface in the form of ‘graphene’. I shall say more about these new arrivals later. Some people regard ‘plasma’, a gas of ions, as a different state of matter, but currently it has little relevance to mainstream chemistry and I shall ignore it.
Gas
A physical chemist is very much at home in a gas. This is the easiest state of matter to describe and to make quantitative predictions about. Indeed, the study of gases could be taken to be yet another springboard of physical chemistry, with Robert Boyle (1627–91) and his investigation of the ‘spring of the air’ in Oxford in about 1660. The study of gases was stimulated further by another feature that was to persist through the centuries, when mankind took to the air in balloons and technological advances depended on further basic research. I have in mind Jacques Charles (1746–1823), his flight in 1783, and the later formulation of his law by another pioneering chemical balloonist, Joseph Louis Gay-Lussac (1778–1850), in 1802.
Boyle, Charles, and Gay-Lussac jointly identified the bulk properties that characterize what we know as a perfect gas (or ‘ideal gas’), essentially the soul of gassiness. Their experiments were crude and the subtle nuances that distinguish one gas from another were washed away in the imprecision of their measurements. That was a good thing at that primitive epoch of science, for they were able to identify simple laws without being distracted by small discrepancies that could be accommodated later. Thus, Boyle established the relation between the volume and the pressure of a gas (V ∝ 1/p) and Charles and Gay-Lussac the relation between the volume and what we now know as the absolute temperature (V ∝ T). To this behaviour in the early 19th century Amedeo Avogadro (1776–1856) added a final contribution, when he hypothesized that equal volumes of gases at the same temperature and pressure contain equal numbers of molecules. In other words, the volume of a gas is proportional to the number of molecules present (V ∝ N). Thus, by combining these observations, was established the ‘perfect gas law’, V ∝ NT/p. Rather astonishingly, the missing universal constant of proportionality in this relation, universal in the sense that it is the same constant for all gases regardless of their identity, is Boltzmann’s constant, k, which I introduced in Chapter 3, and the law is then V = NkT/p. That k occurs is another sign of the underlying ubiquity of the Boltzmann distribution in the description of matter, and I shall explore why it arises in this context shortly.
Comment
The relation V = NkT/p is normally written in the form pV = nRT. The ‘gas constant’ R is related to k by R = NAk, where NA is Avogadro’s constant and n is the amount of substance. I am reluctant to smother the discussion with definitions, except discreetly.
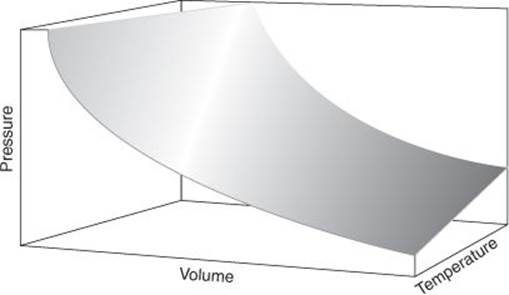
11. The perfect gas equation of state summarizes the allowed states of a perfect gas, a gas in which the molecules neither attract nor repel each other. This surface shows the values of pressure allowed for given values of the volume and temperature of the gas
The perfect gas law, which I will now rearrange into p = NkT/V by multiplying both sides by p/V, is an example of an equation of state, an expression that shows how the pressure is related to other variables under our control, in this case the number of molecules in the sample, the temperature, and the volume it occupies (Figure 11). The perfect gas equation of state is the simplest example and is found to be obeyed by actual gases increasingly well as the pressure is reduced. That is, the perfect gas law is a limiting law in the sense that it is strictly valid only in a certain limit, in this case as p → 0. Actual gases at nonzero pressures are described by more complicated equations of state which are not always known but for which approximate expressions have been established. Happily, though, the perfect gas equation is a very good approximation at normal pressures, and is widely used as a starting point in discussions in physical chemistry.
The existence of the perfect gas equation of state brings me to a very important aspect of physical chemistry: model building. Physical chemists regard empirically established relations (that is, relations between properties that have been established by experiment, such as an equation of state) as a challenge: what molecular interpretation can be devised that accounts for the observed law or relation and then how can it be modified to accommodate deviations from the simple law? In the case of gases, the challenge is to build a model of a perfect gas that agrees with the observed perfect gas equation of state and then to refine the model so that the equations of state of actual gases are obtained. Model building pervades physical chemistry, not just in the description of gases, and I shall give several examples as these chapters unfold. Indeed, some hold that the whole of science is based on building models of physical reality; much of physical chemistry certainly is.
The model of matter that accounts for the perfect gas equation of state is one of the most extraordinary in science: it is essentially ignorance. The kinetic theory of gases accepts that we really don’t know anything about the details of what is happening in a gas except that the molecules are in ceaseless random motion and undergo collisions with one another and the walls of the container. When that model is explored, it turns out that p ∝ N/V, with a constant of proportionality related to the average speed of the molecules in the sample. That mean speed can be calculated from the Boltzmann distribution, which (as I explained in Chapter 3) also arises from ignorance, ignorance about the instantaneous distribution of molecules over their available energy states, in this case the energy states associated with their free motion through space. The missing constant turns out to be simply kT, so we have arrived at the expression p = NkT/V, which is exactly the perfect gas equation of state. From ignorance about the details of what the molecules are doing, we have arrived at an experimentally verified conclusion.
I need to make several points about this model and the conclusions drawn from it. First, it ignores the fact that real molecules interact with one another, attracting each other when they are nearby and repelling one another as soon as they come into contact. Because this contribution has been ignored we can suspect that the deviations that actual gases (so-called ‘real gases’) show from the perfect gas equation of state are due to these interactions. Therefore, to enhance the model and account for deviations from perfect behaviour, these interactions must be incorporated. That is one of the tasks of statistical thermodynamics (Chapter 3) or, if that approach is too challenging, by suggesting ways in which the perfect gas equation of state might be modified. For instance, Johannes van der Waals (1837–1923) proposed a modification in 1873 in which the attractions and repulsions were modelled by two parameters, a and b. One of these parameters, b, is easy to understand: it represents the repulsions that forbid one molecule to occupy the space already occupied by another, and simply reduces the volume available to the gas; thus, the repulsions change p = NkT/V to p = NkT/(V − b). The resulting van der Waals equation of state (which also includes a term proportional to a for the effect of the attractions between molecules) captures many of the properties of real gases and is commonly used by physical chemists to model their behaviour (Figure 12).
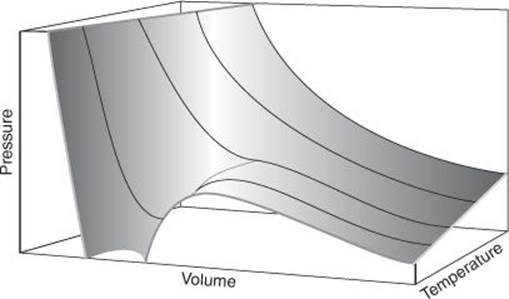
12. When the gas molecules do interact with one another, the pressure depends on the volume in a more complicated way than is shown in Figure 11
A second point is that the model can be developed to deduce some details about the average behaviour of molecules in a gas and so to provide some insight into what is going on in any sample of gas. Insight like that is crucial for understanding the properties of gases and the reactions they undergo. Thus, the Boltzmann distribution can be used to calculate the average speed of the molecules and the fraction of molecules having a specified speed (Figure 13). For reasonably light molecules (such as the major constituents of air, N2 and O2) at room temperature, the molecules are whizzing around at an average speed of about 500 m/s (about 1000 mph). That speed is consistent with what we know about the propagation of sound, the speed of which is about 340 m/s through air: for sound to propagate, molecules must adjust their position to give a wave of undulating pressure, so the rate at which they do so must be comparable to their average speeds.
The kinetic model can be elaborated to predict the average frequency with which molecules collide and the average distance they travel between collisions. To do so, the pointlike molecules of the basic theory are imagined as being surrounded by a sphere of a certain radius that depends on the identity of the molecules, and counting as a ‘hit’ whenever two such spheres come into contact. When this elaboration is worked through, it turns out that a typical N2 or O2 molecule in air makes a collision every nanosecond and travels about 1000 molecular diameters between collisions. To put this scale into perspective: if a molecule is thought of as being the size of a tennis ball, then it travels about the length of a tennis court between collisions. Each molecule makes about a billion collisions a second.
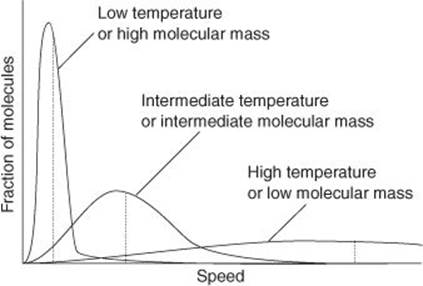
13. The ‘Maxwell–Boltzmann distribution of speeds’ shows the fraction of molecules having a given speed at a specified temperature and can be used to calculate the mean speed of the molecules. Light molecules not only have high average speeds (marked by the dotted lines) but also a high proportion move very rapidly
Physical chemists, in collaboration with physicists, have used the kinetic model to discuss the rate at which properties are transported through gases and the rate of impacts of molecules on surfaces. For instance, they are able to relate the thermal conductivity of a gas, its ability to transport heat down a temperature gradient, to molecular properties. The model is also central to the discussion of the rates at which chemical reactions take place in gases (Chapter 6). It really is quite extraordinary how much information can be extracted from almost complete ignorance.
Liquid
Liquids are a hard nut to crack. Physical chemists have to be interested in them because so much chemistry takes place in them as a medium that it would be a grave omission not to explore and model their properties. The difficulty with understanding them is that they are intermediate between two simple extremes: the chaotic turmoil of molecules in a gas and the rigid arrays of atoms in a solid. Moreover, the most essential liquid to understand, water, is so bizarre that a deep understanding of its properties remains tantalizingly elusive.
There are several aspects of liquids that are open to experimental investigation; they include where the molecules lie at any instant, how fast they are moving, and how easy it is for them to migrate past one another. They can also be studied by monitoring the migration of dissolved substances through them and by observing the way in which they absorb different kinds and colours of electromagnetic radiation. Each experimental method, as is typical of physical chemistry, invites the construction of a model to explain the results. Unlike in a gas, of course, where intermolecular interactions are, to a first approximation, negligible, in liquids, where one molecule jostles against another, they are central to the description.
Even the concept of where molecules lie is problematical, for they are in ceaseless motion, migrating past one another, twisting and turning as they go. To describe the structure, such as it is, of a liquid all we can do is to report the probability that one molecule will be at a certain distance from another. Information of that kind can be obtained experimentally by much the same techniques as are used for solids (X-ray diffraction; see below), and it is found that a typical molecule is surrounded by a shell of other molecules, and then a less well-defined shell surrounds those, and maybe another even more ill-defined shell around those (Figure 14). Water, as in so many respects, is peculiar, as the liquid is found to consist of regions of moderately well-defined ‘ice-like’ regions amid amorphous ‘liquid-like’ regions. The starting point for modelling liquids is to think about how small spheres stack together when shaken together, and the general features of the local structures of liquids are quite well emulated by pots loosely packed with ball bearings.
Physical chemists have several ways of studying the dynamics of molecules in liquids, the characteristics of their motion. The picture to bear in mind is that of a jostling crowd of people; if liquid flow is being studied, then the picture turns into that of a crowd of spectators leaving a packed stadium.
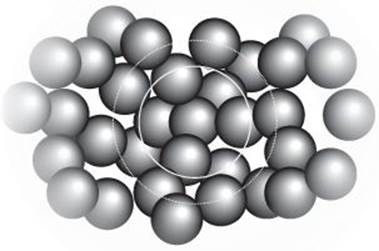
14. A depiction of the structure of a liquid composed of spherical molecules. Each molecule is surrounded by a shell of other molecules, and then a less well-defined shell surrounds those, and maybe another even more ill-defined shell around those. The whole structure is mobile
The classical way of studying the motion of molecules is to measure its ‘viscosity’, its resistance to flow, over a range of temperatures. Viscosities, though, are notoriously difficult to interpret although of considerable practical significance, as in the design of chemical plant where liquids are forced to flow through pipes from place to place. Insight into the motion of molecules themselves is obtained from a variety of techniques. One is neutron scattering: streams of the uncharged cousin of the proton, neutrons, are directed into a liquid sample and bounce off the liquid’s molecules with a variety of energies. By examining those energies a picture can be constructed of the dynamics of the molecules and clusters of cooperating molecules acting in a coordinated manner can be constructed. That other helpful particle, the photon, shot from lasers can also give information of a similar kind.
I discuss nuclear magnetic resonance (NMR) in Chapter 7, but need to mention it here, for it is another technique that can be used to study the motion of molecules in liquids. In particular it can be used to study how molecules rotate in the dense, restrictive environment within a liquid. It is found that in some cases molecules rotate in a series of tiny steps, taking many steps to rotate completely, whereas in some liquids a molecule might swing through a right angle or so at every step.
Physical chemists have been deeply involved in building models of substances dissolved in water and using those dissolved substances to examine the dynamics of molecular motion. Ionic solutes (think sodium chloride in water, with its Na+ and Cl− ions dispersed among the water molecules) are particularly helpful in this respect because the ions can be dragged through the solvent by applying electric fields and the rate at which they move examined by monitoring the resulting electric current. Some initially counterintuitive results are obtained. Thus, it is found that big ions migrate more readily than little ions. That observation led to a picture in which the ions were regarded as being surrounded by water molecules that stuck to them and made them appear bigger because the whole assembly had to migrate through the liquid. Little ions attract water molecules more strongly than big ions (they give rise to a stronger electric field), so with their coating of water molecules masquerade as bigger ions.
This model of ions moving through water was nearly demolished when it was found that hydrogen ions, the smallest ions of all, far from behaving like little ions and moving slowly with a great coating of water molecules, were the most nimble of all! The interpretation gave insight into the behaviour of protons in water, for it was concluded that they didn’t actually move at all. Instead, there is just an effective migration of a proton as neighbouring water molecules shift their ownership of their protons. Thus, in a chain of water molecules like
![]()
a subtle shift in ownership results in
![]()
and the proton effectively and suddenly pops up at the other end of the chain and there is no actual lumbering migration of the original proton.
An early model of the structure of solutions of ions in water is still in use today by physical chemists as a first approximation to the behaviour of ions in solution and in particular their thermodynamic properties. This model was proposed by Peter Debye (1884–1966) and Erich Hückel (1896–1980) in 1923 and, to my mind, is an excellent example of the process of building models as it involves a lot of small steps, each of which is guided by physical insight. The core idea is that a cation (a positive ion) in solution tends to be surrounded by anions (negative ions), and vice versa. Thus, if we think in terms of the time average of their environments, each ion is surrounded by a flimsy ‘ionic atmosphere’ of opposite charge. The interaction of the central ion with its oppositely charged atmosphere lowers its energy and so makes it less thermodynamically active.
Debye and Hückel developed this model quantitatively and deduced what has become known as the Debye–Hückel law in which the energy lowering is proportional to the square root of the concentration of ions present. In fact, theirs is another example of the limiting laws characteristic of physical chemistry: in this case, the approximations made in the course of its development mean that it is strictly valid only in the limit of zero concentration. Despite that limitation, the law is a good starting point for more sophisticated theories of ionic solutions.
Solid
Physical chemistry is on much firmer ground with solids. Here, instead of motion being paramount (as in gases), motion constrained but important (as in liquids), to a first approximation there is no motion at all for all the atoms of a solid can be regarded as in fixed positions. Motion does survive in solids, but it is largely just the quivering of molecules in place as they vibrate, and initially can be ignored.
The principal experimental technique for the determination of the structures of solids is X-ray diffraction (or ‘X-ray crystallography’). However, I have to admit that so central is an understanding of the arrangement of atoms in molecules to molecular biology and inorganic chemistry, that molecular biologists and inorganic chemists probably regard X-ray diffraction as their own; indeed, I think it true to say that physical chemists on the whole are happy to let slip the technique from their intellectual grasp but keep a tight grip on its conclusions. Their conclusions overlap with the interests of materials scientists, for here, as in biology, structure is allied closely with function.
That—the distance that physical chemists largely keep from the technique itself—being so, I will not dwell on it for more than a sentence or so, and focus instead on what it reveals. In short, X-ray diffraction makes use of the fact that electromagnetic radiation (which includes X-rays) consists of waves that can interfere with one another and give rise to regions of enhanced and diminished intensity. This so-called ‘diffraction pattern’ is characteristic of the object in the path of the rays, and mathematical procedures can be used to interpret the pattern in terms of the object’s structure. Diffraction occurs when the wavelength of the radiation is comparable to the dimensions of the object. X-rays have wavelengths comparable to the separation of atoms in solids, so are ideal for investigating their arrangement.
Crystals, as was speculated long ago by René Haüy (1743–1822) in about 1784, following similar speculations by Johannes Kepler in 1611 and Robert Hooke in 1665, and confirmed by X-ray diffraction in the 20th century, consist of serried ranks of atoms in uniform arrays. The most primitive model for accounting for the structures of crystals of elemental metals (silver, copper, iron, etc.) is to regard each atom as a hard sphere and to consider how these spheres may be stacked together in layers to reproduce the entire crystal. Many metals have ‘close-packed’ structures in which the spheres pack together in the most efficient way possible (Figure 15). One consequence is that metals are dense materials as so much space is occupied by their closely packed atoms.
Ionic solids present a double problem: cations and anions have different radii and opposite charges. The lowest energy arrangement is obtained only if cations are surrounded by anions, and vice versa (the sodium chloride structure shown in Figure 3 (Chapter 1) is an example). This requirement puts a severe restraint on the possible arrangements of the ions and the dense close packing characteristic of metals is never achieved. Largely for that reason, ionic solids are typically less dense than metals. If those two problems are not enough, there are more: many solids have different numbers of cations and anions (calcium chloride, for instance, CaCl2), and in some solids covalent bonding, with its characteristic directional properties, distorts the arrangements expected on the basis of simple sphere-packing. If those four problems, at this stage, are also not enough, then an additional one is that in many cases the ions are far from spherical (sulfate ions, SO42−, for instance, are tetrahedral, and there are many more elaborate instances), and all manner of peculiar shapes need to be stacked together in uniform arrays with no overall net electric charge.
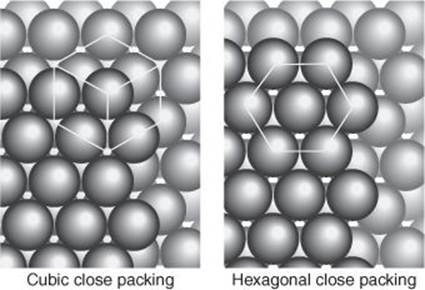
15. There are two ways of packing identical uncharged spheres together, one gives a ‘cubic’ arrangement and the other gives a ‘hexagonal’ arrangement. Copper is an example of the former, zinc of the latter. The type of packing affects the mechanical properties of the metal. (The lines indicating cubic symmetry join spheres on three layers)
Physical chemists have been awed but not daunted by these problems. They have appealed extensively to computers to assemble all manner of crystals from elaborate shapes, building into the software models of how the different components interact with one another and setting the computer to search for the arrangements of lowest energy. This type of study has proved particularly fruitful for the development of the elaborate solids now in vogue as catalysts. These porous solids often have wide channels between blocks of ions to allow the penetration and escape of reactants. The channels effectively increase the surface area enormously by making the reactive hotspots in the interior of the solid accessible to reactant molecules.
Chemical reactivity is not the only aspect of solids in which physical chemists take an interest. Here their interests merge into those of physicists and materials scientists, who will have techniques for computing the electrical, optical, and mechanical properties of even quite complex solids and finding, in collaboration with physical and inorganic chemists (and increasingly with organic chemists), how to enhance the properties in which they are interested. One area that immediately springs to mind where fruitful collaboration lies is the development of the ceramic materials that show the extraordinary property of superconductivity, the ability to conduct electricity without resistance, at temperatures not enormously far below room temperature. Another is the development of electrode and electrolyte materials for the efficient production of electricity by modern batteries.
Intermediate states of matter
I suspect that almost all classifications have frayed edges where an entity exists that is neither fish nor fowl and perhaps a little of both. Frayed frontiers are often regions of considerable interest, where interesting properties lurk and from where new understanding and technologies spring. There are two such frontiers that interest physical chemists, one where liquid meets gas and the other where liquid meets solid.
Liquid meets gas in an interesting way at and around the ‘critical point’ of a substance. To appreciate the significance of that point, imagine a liquid and its vapour in a sealed, rigid container; we shall imagine it to be transparent so that we can see what is happening. As it is heated, the liquid evaporates and consequently the density of the vapour above it increases. There will come a stage when the density of the vapour matches that of the liquid and the surface between them disappears. This is the critical point. The substance in the vessel is neither liquid nor gas but a supercritical fluid. A supercritical fluid has a density comparable to that of a liquid but fills the container it occupies, like a gas.
Supercritical fluids are seriously interesting not only for their intrinsic properties but for their potential as solvents. Among the most heavily studied is supercritical carbon dioxide, scCO2, as a non-noxious, environmentally benign solvent for many organic compounds and which can be removed without leaving any contamination simply by allowing it to vaporize. Supercritical carbon dioxide occurs at around 31°C at pressures of around 73 atm (1 atm is normal atmospheric pressure at sea level).
Supercritical carbon dioxide is at the centre of attention for an increasing number of solvent-based processes. The transport properties of any supercritical fluid (its viscosity and thermal conductivity, for instance) depend strongly on its density, which in turn is sensitive to the pressure and temperature: the density of supercritical carbon dioxide can be adjusted from a gas-like 0.1 g/cm3 to a liquid-like 1.2 g/cm3. The solubility of a solute is strongly dependent on the density of the supercritical fluid, so small increases in pressure, particularly close to the critical point, can have very large effects on solubility.
Physical chemists are central to the establishment of the conditions under which a substance can be regarded as supercritical and the exploration of the properties of such fluids and the processes that occur in them is a potentially vigorous area of research. Water itself goes supercritical, but the conditions (around 374°C and 218 atm) are less favourable than for carbon dioxide. Nevertheless, it is a tantalisingly interesting domain for experimental and theoretical exploration.
Physical chemists have become very interested in ‘soft matter’ (also called ‘complex fluids’) which lies in character between hard solids and liquids. Many polymeric materials are of this kind, as are surface coatings, adhesives, foams, emulsions, and many biological materials.
Liquid crystals, one form of soft matter, are familiar from electronic displays. They form a so-called mesophase state of matter with properties related to those of both liquids and solids. To see liquid crystals in context, recall that in a solid there is long-range order: serried rank follows serried rank right to the edge of a crystal. In contrast, a liquid has only short-range order: there is predictable structure of the first shell of molecules around a central molecule, less predictable in the next shell, and barely any structure for shells of molecules much further away. In a liquid crystal there is long-range order in one or two dimensions but only short-range order in the remaining dimension. For instance, in one type of liquid crystal, the long thin molecules that lie in a plane in a regular pattern, but the parallel planes above and below lie in orientations that show little relation to that central plane (Figure 16).
Liquid crystals have interesting optical properties, as can be inferred from their use in optical displays. Unlike liquids, they also have viscous and mechanical properties that depend on direction. Physical chemists are closely involved in the development of various types of liquid crystal properties and establishing the dynamical properties of the molecules within and between the layers. This type of examination also impinges on biology, for liquid crystals are very similar to the membranes that encapsulate the cells of organisms, including our own.

16. In one form of a liquid crystal (the ‘nematic’ phase) molecules lie in orderly ranks in each plane but neighbouring planes adopt different orientations
One great class of materials that contribute to the class of ‘soft matter’ substances are the polymers, such as polyethylene, nylon, and all their elaborations. Polymers are a great romping ground for physical chemists as their structures and properties vary so widely. They vary mechanically from the rigid to the runny, electrically from insulator to conductor, and optically too. Some of their properties are modified by the application of electric fields. They represent challenges for their characterization, such as their molecular weights (see the next chapter for a comment), the spread of molecular weights in a sample, the manner in which they might roll up into tight balls or unroll into gangly strands, and the length and conformations of the polymer chains.
The current challenge
Soft polymeric materials are beginning to provide physical chemists with the opportunity to modify materials, including the colours of fabrics, with electric fields, and are offering great opportunities for imaginative investigation both experimental and theoretical. Soft matter presents challenges about structure and its relation to properties, especially the response of samples to sudden changes in the conditions, including mechanical impact. To investigate responses such as these it is essential to build models of the dynamical behaviour of the molecular components and then to convert the theoretical description of that behaviour into a description of the behaviour of bulk samples.
Hard matter also presents challenges, especially when it has fascinating optical and electronic properties, such as superconductivity, and has potential for the storage and manipulation of information. Almost literally at the interface of solids lies graphene, the closest physical chemists come to inhabiting Flatland. Graphite itself is stacks of layers of carbon atoms in a chicken-wire arrangement: these layers slide over each other when impurities are present, which is why impure graphite can be used as a lubricant. Graphene is one of these sheets in isolation. The remarkable strength of this material, and its intriguing electrical properties are both of great interest and are beginning to be studied by physical chemists who now are confronted by the challenges represented by this essentially two-dimensional material. Similarly intriguing materials are aperiodic crystals, which though built from atoms that are packed together closely, do not have the essentially infinite order characteristics of conventional crystals.