Physical Chemistry: A Very Short Introduction (2014)
Chapter 6. Changing the identity of matter
A great deal of chemistry is concerned with changing the identity of matter by the deployment of chemical reactions. It should hardly be surprising that physical chemistry is closely interested in the processes involved and that it has made innumerable contributions to our understanding of what goes on when atoms exchange partners and form new substances. Physical chemists are interested in a variety of aspects of chemical reactions, including the rates at which they take place and the details of the steps involved during the transformation. The general field of study of the rates of reactions is called chemical kinetics. When the physical chemist gets down to studying the intimate details of the changes taking place between atoms, chemical kinetics shades into chemical dynamics.
Chemical kinetics is a hugely important aspect of physical chemistry for it plays a role in many related disciplines. For instance, it is important when designing a chemical plant to know the rates at which intermediate and final products are formed and how those rates depend on the conditions, such as the temperature and the presence of catalysts. A human body is a network of myriad chemical reactions that are maintained in the subtle balance we call homeostasis, and if a reaction rate runs wild or slows unacceptably, then disease and death may ensue. The observation of reaction rates and their dependence on concentration of reactants and the temperature gives valuable insight into the steps by which the reaction takes place, including the role of a catalyst, and how the rates may be optimized. Chemical dynamics deepens this understanding by putting individual molecular changes under its scrutiny.
Then there are different types of chemical reactions other than those achieved simply by mixing and heating. Some chemical reactions are stimulated by light, and physical chemists play a central role in the elucidation of these ‘photochemical reactions’ in the field they call photochemistry. There is no more important photochemical reaction than the photosynthesis that captures the energy of the Sun and stands at the head of the food chain, and physical chemists play a vital role in understanding its mechanism and devising ways to emulate it synthetically. Then there is the vital collaboration of electricity and chemistry in the form of electrochemistry. The development of this aspect of chemistry is central to modern technology and the deployment of electrical power.
Spontaneous reaction
The first contribution to understanding chemical reactions that physical chemists make is to identify, or at least understand, the spontaneous direction of reaction. (Recall from Chapter 2 that ‘spontaneous’ has nothing to do with rate.) That is, we need to know where we are going before we start worrying about how fast we shall get there. This is the role of chemical thermodynamics, which also provides a way to predict the equilibrium composition of the reaction mixture, the stage at which the reaction seems to come to a halt with no tendency to change in either direction, towards more products or back to reactants. Remember, however, that chemical equilibria are dynamic: although change appears to have come to an end, the forward and reverse reactions are continuing, but now at matching rates. Like the equilibria we encountered in Chapter 5, chemical equilibria are living, responsive conditions.
The natural direction of a chemical reaction corresponds to time’s usual arrow: the direction of increasing entropy of the universe. As explained in Chapter 2, provided the pressure and temperature are held constant, attention can be redirected from the entire universe to the flask or test-tube where their reaction is taking place by focusing instead on the Gibbs energy of the reaction system. If at a certain stage of the reaction the Gibbs energy decreases as products are formed, then that direction is spontaneous and the reaction has a tendency to go on generating products. If the Gibbs energy increases, then the reverse reaction is spontaneous and any products present will have a tendency to fall apart and recreate the reactants. If the Gibbs energy does not change in either direction, then the reaction is at equilibrium.
These features can all be expressed in terms of the chemical potential of each participant of the reaction, the concept introduced in Chapter 5 in connection with physical change. The name ‘chemical potential’ now fulfils its significance. Thus, the chemical potentials of the reactants effectively push the reaction towards products and those of the products push it back towards reactants. The reaction is like a chemical tug-of-war with the chemical potentials the opponents pushing rather than pulling: stalemate is equilibrium. Physical chemists know how the chemical potentials change as the composition of the reaction mixture changes, and can use that information to calculate the composition of the mixture needed for the chemical potentials of all the participants to be equal and the reaction therefore to be at equilibrium. I have already explained (in Chapter 3) that the mixing of the reactants and products plays a crucial role in deciding where that equilibrium lies.
Physical chemists don’t deduce the equilibrium composition directly. The thermodynamic expressions they derive are in terms of a combination of the concentrations of the reactants and products called the reaction quotient, Q. Broadly speaking, the reaction quotient is the ratio of concentrations, with product concentrations divided by reactant concentrations. It takes into account how the mingling of the reactants and products affects the total Gibbs energy of the mixture. The value of Q that corresponds to the minimum in the Gibbs energy can be found, and then the reaction tug-of-war is in balance and the reaction is at equilibrium. This particular value of Q is called the equilibrium constant and denoted K. The equilibrium constant, which is characteristic of a given reaction and depends on the temperature, is central to many discussions in chemistry. When K is large (1000, say), we can be reasonably confident that the equilibrium mixture will be rich in products; if K is small (0.001, say), then there will be hardly any products present at equilibrium and we should perhaps look for another way of making them. If K is close to 1, then both reactants and products will be abundant at equilibrium and will need to be separated.
The equilibrium constant is, as this discussion implies, related to the change in Gibbs energy that accompanies the reaction, and the relation between them is perhaps one of the most important in chemical thermodynamics. It is the principal link between measurements made with calorimeters (which, as explained in Chapter 2, are used to calculate Gibbs energies) and practical chemistry, through a chemist’s appreciation of the significance of equilibrium constants for understanding the compositions of reaction mixtures.
Comment
This important equation is ![]() ln K, where
ln K, where ![]() is the change in Gibbs energy on going from pure reactants to pure products and T is the temperature; R is the gas constant, R = NAk.
is the change in Gibbs energy on going from pure reactants to pure products and T is the temperature; R is the gas constant, R = NAk.
Equilibrium constants vary with temperature but not, perhaps surprisingly, with pressure. The hint that they depend on temperature comes from remembering that the Gibbs energy includes the temperature in its definition (G = H − TS). Jacobus van’t Hoff, who was mentioned in Chapter 5 in relation to osmosis, discovered a relation between the sensitivity of the equilibrium constant to temperature and the change in enthalpy that accompanies the reaction. In accord with Le Chatelier’s principle about the self-correcting tendency of equilibria exposed to disturbances, van’t Hoff’s equation implies that if the reaction is strongly exothermic (releases a lot of energy as heat when it takes place), then the equilibrium constant decreases sharply as the temperature is raised. The opposite is true if the reaction is strongly endothermic (absorbs a lot of energy as heat). That response lay at the root of Haber and Bosch’s scheme to synthesize ammonia, for the reaction between nitrogen and hydrogen is strongly exothermic, and although they needed to work at high temperatures for the reaction to be fast enough to be economical, the equilibrium constant decreased and the yield would be expected to be low. How they overcame this problem I explain below.
Reaction rate
As I have emphasized, thermodynamics is silent on the rates of processes, including chemical reactions, and although a reaction might be spontaneous, that tendency might be realized so slowly in practice that for all intents and purposes the reaction ‘does not go’. The role of chemical kinetics is to complement the silence of thermodynamics by providing information about the rates of reactions and perhaps suggesting ways in which rates can be improved.
What is meant by the ‘rate’ of a chemical reaction? Rate is reported by noting the change in concentration of a selected component and dividing it by the time that change took to occur. Just as the speed of a car will change in the course of a journey, so in general does the rate of a reaction. Therefore, to get the ‘instantaneous rate’ of a reaction (like the actual speed of a car at any moment), the time interval between the two measurements of concentration needs to be very small. There are technical ways of implementing this procedure that I shall not dwell on. Typically it is found that the rate of a reaction decreases as it approaches equilibrium.
That the instantaneous rate (from now on, just ‘rate’) depends on concentration leads to an important conclusion. It is found experimentally that many reactions depend in a reasonably simple way on the concentrations of the reactants and products. The expression describing the relation between the rate and these concentrations is called the rate law of the reaction. The rate law depends on one or more parameters, which are called rate constants. These parameters are constant only in the sense that they are independent of the concentrations of the reactants and products: they do depend on the temperature and typically increase with increasing temperature to reflect the fact that most reactions go faster as the temperature is increased.
Some rate laws are very simple. Thus it is found that the rates of some reactions are proportional to the concentration of the reactants and others are proportional to the square of that concentration. The former are classified as ‘first-order reactions’ and the latter as ‘second-order reactions’. The advantage of this classification, like all classifications, is that common features can be identified and applied to reactions in the same class. In the present case, each type of reaction shows a characteristic variation of composition with time. The concentration of the reactant in a first-order reaction decays towards zero at a rate determined by its rate constant; that of a second-order reactions decays to zero too, but although it might start of at the same rate, the concentration of reactant takes much longer to reach zero. An incidental point in this connection is that many pollutants in the environment disappear by a second-order reaction, which is why low concentrations of them commonly persist for long periods.
One of the points to note is that it isn’t possible to predict the order of the reaction just by looking at its chemical equation. Some very simple reactions have very complicated rate laws; some that might be expected to be first order (because they look as though a molecule is just shaking itself apart) turn out to be second order, and some that might be suspected to be second order (for example, they look as though they take place by collisions of pairs of molecules) turn out to be first order.
One aim of chemical kinetics is to try to sort out this mess. To do so, a reaction mechanism (a sequence of steps involving individual molecules) is proposed that is intended to represent what is actually happening at a molecular level. Thus, it might be supposed that in one step two molecules collide and that one runs off with much more energy than it had before, leaving the other molecule depleted in energy. Then in the second step that excited molecule shakes itself into the arrangement of atoms corresponding to the product. The rate laws for these individual steps (and these laws can be written down very simply: in this case the first step is second order and the second step is first order) are combined to arrive at the rate law for the overall reaction. If the rate law matches what is observed experimentally, then the proposed mechanism is plausible; if it doesn’t match, then a different mechanism has to be proposed. The problem is that even if the overall rate law is correct, there might be other mechanisms that lead to the same rate law, so other evidence has to be brought to bear to confirm that the mechanism is indeed correct. In that respect, the establishment of a reaction mechanism is akin to the construction of proof of guilt in a court of law.
In some cases it is possible to identify a step that controls the rate of the entire overall process. Such a step is called the rate-determining step. Many steps might precede this step, but they are fast and do not have a significant influence on the overall rate. A mechanism with a rate-determining step has been likened to having six-lane highways linked by a one-lane bridge.
Reaction rate and temperature
Most reactions go faster when the temperature is raised. The reason must be found in the rate constants in the rate law, for only they depend on temperature. The specific temperature dependence that is typical of rate constants was identified by Svante Arrhenius (1859–1927) who proposed in 1889 that their dependence on temperature could be summarized by introducing two parameters, the more interesting of which became known as the activation energy of the reaction: reactions with high activation energies proceed slowly at low temperatures but respond sharply to changes of temperature.
Comment
The Arrhenius expression is ![]() , where kr is the rate constant and Ea is the activation energy; A is the second parameter.
, where kr is the rate constant and Ea is the activation energy; A is the second parameter.
The simplest explanation of the so-called ‘Arrhenius parameters’ is found in the collision theory of gas-phase reactions. In this theory, the rate of reaction depends on the rate at which molecules collide: that rate can be calculated from the kinetic theory of gases, as explained in Chapter 4. However, not all the collisions occur with such force that the atoms in the colliding molecules can rearrange into products. The proportion of high-energy collisions can be calculated from the Boltzmann distribution (Chapter 3). Then, the expression developed by Arrhenius is obtained if it is supposed that reaction occurs only if the energy of the collision is at least equal to the activation energy (Figure 21). Thus, the activation energy is now interpreted as the minimum energy required for reaction. Increasing the temperature increases the proportion of collisions that have at least this energy, and accordingly the reaction rate increases.
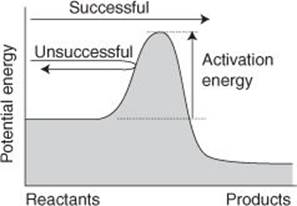
21. The activation energy is the minimum energy required for reaction and can be interpreted as the height of a barrier between the reactants and products: only molecules with at least the activation energy can pass over it
Gas-phase reactions are important (for instance, for accounting for the composition of atmospheres as well as in a number of industrial processes), but most chemical reactions take place in solution. The picture of molecules hurtling through space and colliding is not relevant to reactions in solution, so physical chemists have had to devise an alternative picture, but one that still accounts for the Arrhenius parameters and particularly the role of the activation energy.
The picture that physical chemists have of reactions in solution involves two processes. In one, the reactants jostle at random in the solution and in due course their paths may bring them into contact. They go on jostling, and dance around each other before their locations diverge again and they go off to different destinies. The second process occurs while they are dancing around each other. It may be that the ceaseless buffeting from the solvent molecules transfers to one or other of the reactant molecules sufficient energy—at least the activation energy—for them to react before the dance ends and they separate.
In a ‘diffusion-controlled reaction’ the rate-determining step is the jostling together, the diffusion through the solution, of the reactants. The activation energy for the reaction step itself is so low that reaction takes place as soon as the reactants meet. In this case the metaphorical narrow bridge precedes the six-lane highway, and once the reactants are over it the journey is fast. The diffusion together is the rate-determining step. Diffusion, which involves molecules squirming past one another, also has a small activation energy, so Arrhenius-like behaviour is predicted. In an ‘activation-controlled reaction’ the opposite is true: encounters are frequent but most are sterile because the actual reaction step requires a lot of energy: its activation energy is high and constitutes the rate-determining step. In this case, the six-lane highway leads up to the narrow bridge.
It is much harder to build a model of the reaction step in solution than it is to build the model for gas-phase reactions. One approach is due to Henry Eyring (1901–61), who provides another scandalous example of a Nobel Prize deserved but not awarded. In his transition-state theory, it is proposed that the reactants may form a cluster of atoms before falling apart as the products. The model is formulated in terms of statistical thermodynamic quantities, with the ubiquitous Boltzmann distribution appearing yet again. Transition-state theory has been applied to a wide variety of chemical reactions, including those responsible for oxidation and reduction and electrochemistry. The principal challenge is to build a plausible model of the intermediate cluster and to treat that cluster quantitatively.
Catalysis
A catalyst is a substance that facilitates a reaction. The Chinese characters for it make up ‘marriage broker’, which conveys the sense very well. Physical chemists are deeply involved in understanding how catalysts work and developing new ones. This work is of vital economic significance, for just about the whole of chemical industry throughout the world relies on the efficacy of catalysts. Living organisms owe a similar debt, for the protein molecules we call enzymes are highly effective and highly selective catalysts that control almost every one of the myriad reactions going on inside them and result in them ‘being alive’.
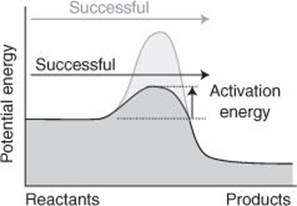
22. A catalyst provides an alternative reaction pathway with a low activation energy
All catalysts work by providing a different, faster pathway for a reaction. In effect, they provide a pathway with an activation energy that is lower than in the absence of the catalyst (Figure 22). Most industrial catalysts work by providing a surface to which reactant molecules can attach and in the process of attachment perhaps be torn apart. The resulting fragments are ripe for reaction, and go on to form products. For physical chemists to contribute to the development of catalysts, they need to understand the structures of surfaces, how molecules attach to (technically: ‘adsorb on’) them, and how the resulting reactions proceed. They employ a variety of techniques, with computation used to model the reaction events and observation used to gather data. A surge in their ability to examine surfaces took place a few years ago with the development of the techniques I describe in Chapter 7 that provide detailed images of surfaces on an atomic scale.
Biological catalysts, enzymes, require different modes of investigation, but with computation still a major player and physical chemists in collaboration with molecular biologists, biochemists, and pharmacologists. Nowhere in chemistry is shape more central to function than in the action of enzymes, for instead of catalysis occurring on a reasonably flat and largely featureless surface, an enzyme is a highly complex structure that recognizes its prey by shape, accepts it into an active region of the molecule, produces change, and then delivers the product on to the next enzyme in line. Disease occurs when an enzyme ceases to act correctly because its shape is modified in some way and becomes unable to recognize its normal prey and perhaps acts on the wrong molecule or when its active site is blocked by an intrusive molecule and it fails to act at all. The recognition of the important role of shape provides a hint about how a disease may be cured, for it may be possible to build a molecule that can block the action of an enzyme that has gone wild or reawaken a dormant enzyme. Physical chemists in collaboration with pharmacologists make heavy users of computers to model the docking of candidate molecules into errant active sites.
Photochemistry
Photochemistry plays an important role in physical chemistry not only because it is so important (as in photosynthesis) but also because the use of light to stimulate a reaction provides very precise control over the excitation of the reactant molecules. The field has been greatly influenced by the use of lasers with their sharply defined frequencies and their ability to generate very short pulses of radiation. The latter enables the progress of reactions to be followed on very short timescales, with current techniques easily dealing with femtosecond phenomena (1 fs = 10−15 s) and progress being made with attosecond phenomena too (1 as = 10–18 s), where chemistry is essentially frozen into physics.
The idea behind photochemistry is to send a short, sharp burst of light into a sample and to watch what happens. There are physical and chemical consequences.
The physical consequences are the emission of light either as fluorescence or as phosphorescence. The two forms of emission are distinguished by noting whether the emission persists after the illuminating source is removed: if emission ceases almost immediately it is classified as fluorescence; if it persists, then it is phosphorescence. Physical chemists understand the reasons for the difference in this behaviour, tracing it to the way that the energetically excited molecules undergo changes of state. In fluorescence, the excited state simply collapses back into the ground state. In phosphorescence, the excited molecule switches into another excited state that acts like a slowly leaking reservoir.
Fluorescence is eliminated if another molecule diffuses up to the excited molecule and kidnaps its excess energy. This ‘quenching’ phenomenon is used to study the motion of molecules in solution, for by changing the concentration of the quenching molecule and watching its impact on the intensity of the fluorescence inferences can be made about what is taking place in the solution. Moreover, the decay of the fluorescence after the stimulating illumination is extinguished is not instantaneous, and the rate of decay, though rapid, can be monitored with or without quenching molecules present and information about the rates of the various processes taking place thereby deduced.
The chemical consequences of photoexcitation stem from the fact that the excited molecule has so much energy that it can participate in changes that might be denied to its unexcited state. Thus, it might be so energy-rich that it shakes itself apart into fragments. These fragments might be ‘radicals’ which are molecules with an unpaired electron. Recall from Chapter 1 that a covalent bond consists of a shared pair of electrons, which we could denote A:B. If a molecule falls apart into fragments, one possibility is that the electron pair is torn apart and two radicals, in this case A· and ·B, are formed. Most radicals are virulent little hornets, and attack other molecules in an attempt to re-form an electron pair. That attack, if successful, leads to the formation of more radicals, and the reaction is propagated, sometimes explosively. For instance, hydrogen and chlorine gases can be mixed and cohabit in a container without undergoing change, but a flash of light can stimulate an explosive reaction between them.
In some cases the photoexcited molecule is just an intermediary in the sense that it does not react itself but on collision with another molecule passes on its energy; the recipient molecule then reacts. Processes like this, including the radical reactions, are enormously important for understanding the chemical composition of the upper atmosphere where molecules are exposed to energetic ultraviolet radiation from the Sun. The reactions are built into models of climate change and the role of pollutants in affecting the composition of the atmosphere, such as the depletion of ozone (O3).
Photochemical processes can be benign. There is no more benign photochemical process than photosynthesis, in which photochemical processes capture the energy supplied by the distant Sun and use it to drive the construction of carbohydrates, thus enabling life on Earth. Physical chemists are deeply involved in understanding the extraordinary role of chlorophyll and other molecules in this process and hoping to emulate it synthetically in what would be a major contribution to resolving our energy problems on Earth and wherever else in due course we travel. Meanwhile, physical chemists also contribute to understanding the photovoltaic materials that are a temporary but important approximation to the power of photosynthesis and make use of the excitation of electrons in inorganic materials.
Electrochemistry
Another major contribution that physical chemistry makes to the survival of human societies and technological advance is electrochemistry, the use of chemical reactions to generate electricity (and the opposite process, electrolysis). A major class of chemical reactions of interest in electrochemistry consists of redox reactions, and I need to say a word or two about them.
In the early days of chemistry, ‘oxidation’ was simply reaction with oxygen, as in a combustion reaction. Chemists noticed many similarities to other reactions in which oxygen was not involved, and came to realize that the common feature was the removal of electrons from a substance. Because electrons are the glue that holds molecules together, in many cases the removal of electrons resulted in the removal of some atoms too, but the core feature of oxidation was recognized as electron loss.
In similar former days, ‘reduction’ was the term applied to the extraction of a metal from its ore, perhaps by reaction with hydrogen or (on an industrial scale in a blast furnace) with carbon. As for oxidation, it came to be recognized that there were common features in other reactions in which metal ores were not involved and chemists identified the common process as the addition of electrons.
Oxidation is now defined as electron loss and reduction is defined as electron gain. Because electron loss from one species must be accompanied by electron gain by another, oxidation is always accompanied by reduction, and their combinations, which are called ‘redox reactions’, are now recognized as the outcome of electron transfer, the transfer perhaps being accompanied by atoms that are dragged along by the migrating electrons.
The core idea of using a chemical reaction to produce electricity is to separate reduction from oxidation so that the electrons released in the oxidation step have to migrate to the location where reduction takes place. That migration of electrons takes place through a wire in an external circuit and constitutes an electric current driven by the spontaneous reaction taking place in the system (Figure 23). All batteries operate in this way, with the electrodes representing the gateways through which the electrons enter or leave each cell. Electrolysis is just the opposite: an external source pulls electrons out of the cell through one electrode and pushes them back into the cell through another, so forcing a non-spontaneous redox reaction to occur.
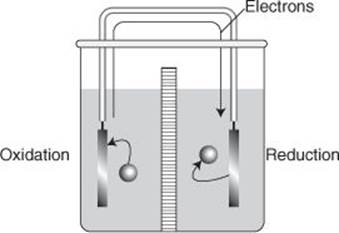
23. In an electrochemical cell, the sites of oxidation (electron loss) and reduction (electron gain) are separated, and as the spontaneous reaction proceeds electrons travel from one electrode to the other
There are ‘academic’ and ‘industrial’ applications of electrochemistry. I pointed out in Chapter 2 that the change in Gibbs energy during a reaction is an indication of the maximum amount of non-expansion work that the reaction can do. Pushing electrons through a circuit is an example of non-expansion work, so the potential difference that a cell can generate (its ‘voltage’), a measure of the work that the cell can do, is a way of determining the change in Gibbs energy of a reaction. You can now see how a thermodynamicist views a ‘flat’ battery: it is one in which the chemical reaction within it has reached equilibrium and there is no further change in Gibbs energy. Physical chemists know how the Gibbs energy is related to the concentrations of the reactants and products (through the reaction quotient, Q), and can adapt this information to predict the voltage of the cell from its composition. If you recall from Chapter 2, I remarked that thermodynamics provides relations, sometimes unexpected ones, between disparate properties. Electrochemists make use of this capability and from how the voltage changes with temperature are able to deduce the entropy and enthalpy changes due to the reaction, so electrochemistry contributes to thermochemistry.
Electrochemists are also interested in the rate at which electrons are transferred in redox reactions. An immediate application of understanding the rates at which electrons are transferred into or from electrodes is to the improvement of the power, the rate of generation of electrical energy, that a cell can produce. But the rates of electron transfer also have important biological implications, because many of the processes going on inside organisms involve the transfer of electrons. Thus, if our brains are operating at 100 watts and there are potential differences within us amounting to about 10 volts, then we are 10 ampere creatures. The understanding of the rate of electron transfer between biologically important molecules is crucial to an understanding of the reactions, for instance, that constitute the respiratory chain, the deployment of the oxygen that we breathe and which drives all the processes within us.
The industrial applications of electrochemistry are hugely important, for they include the development of light, portable, powerful batteries and the development of ‘fuel cells’. A fuel cell is simply a battery in which the reactants are fed in continuously (like a fuel) from outside, rather than being sealed in once and for all at manufacture. Physical chemists contribute to the improvement of their efficiencies by developing new electrode and electrolyte materials (the electrolyte is the medium where the redox reactions take place). Here lie opportunities for the union of electrochemistry and photochemistry, for some cells being developed generate electricity under the influence of sunlight rather than by using a physical fuel.
The metal artefacts fabricated at such expense (of cash and energy) crumble in the face of Nature and the workings of the Second Law. Corrosion, the electrochemical disease of metal, destroys and needs to be thwarted. Electrochemists seek an understanding of corrosion, for it is a redox reaction and within their domain, and through that understanding look for ways to mitigate it and thereby save societies huge sums.
Chemical dynamics
Chemical dynamics is concerned with molecular intimacies, where physical chemists intrude into the private lives of atoms and watch in detail the atomic processes by which one substance changes into another.
One of the most important techniques for discovering what is happening to individual molecules when they react is the molecular beam. As its name suggests, a molecular beam is a beam of molecules travelling through space. It is just not any old beam: the speeds of the molecules can be selected in order that the kinetic energy they bring to any collision can be monitored and controlled. It is also possible to select their vibrational and rotational states and to twist them into a known orientation. In a molecular beam apparatus, one beam impinges on target molecules, which might be a puff of gas or might be another beam. The incoming beam is scattered by the collision or, if the impacts are powerful enough, its molecules undergo reactions and the products fly off in various directions. These products are detected and their states determined. Thus, a very complete picture can be constructed of the way that molecules in well-defined states are converted into products in similarly well-defined states.
Molecular dynamics is greatly strengthened by its alliance with computational chemistry. As two molecules approach each other, their energy changes as bonds stretch and break, they bend, and new bonds are formed. The calculation of these energies is computationally very demanding but has been achieved in a number of simple cases. It results in a landscape of energies known as a potential-energy surface. When molecules travel through space they are, in effect, travelling across this landscape. They adopt paths that can be calculated either by using Newton’s laws of motion or, in more sophisticated and appropriate treatments, by using the laws of quantum mechanics (that is, by solving Schrödinger’s equation).
Close analysis of the paths that the molecules take through the landscape gives considerable insight into the details of what takes place at an atomic level when a chemical reaction occurs. Thus it may be found that reaction is more likely to ensue if one of the molecules is already vibrating as it approaches its target rather than just blundering into it with a lot of translational energy. Reaction might also be faster if a molecule approaches the target from a particular direction, effectively finding a breach in its defensive walls.
Physical chemists need to forge a link between this very detailed information and what happens on the scale of bulk matter. That is, they need to be able to convert the trajectories that they compute across the potential-energy landscape into the numerical value of a rate constant and its dependence on temperature. This bridge between the microscopic and the macroscopic can be constructed, and there is increasing traffic across it.
The current challenge
The challenge that confronts the physical chemists who experiment with molecular beams and their intellectual compatriots who compute the potential energy surfaces on which the interpretation of the reactive and non-reactive scattering depend is to find ways to transfer their techniques and conclusions into liquids, the principal environment of chemical reactions. Although there is little hope of doing so directly (although the continuing massive advances in computing power rule nothing out), the detailed knowledge obtained from the gas phase adds to our insight about what is probably happening in solution and generally enriches our understanding of chemistry.
At an experimental level perhaps nothing is more important to the development of energy-hungry civilization than the role that physical chemistry plays in the development of the generation and deployment of electric power through photochemistry and the improvement of fuel cells and portable sources. A major part of this application is the development of an understanding of surface phenomena, seeking better catalysts, developing better electrode materials, and exploring how photochemical activity can be enhanced. Nature has much to teach in this connection, with clues obtained from the study of photosynthesis and the action of enzymes.
Molecular beams are capable of providing exquisite information about the details of reaction events and laser technology, now capable of producing extraordinarily brief flashes of radiation, permits reaction events to be frozen in time and understood in great detail.
Classical kinetic techniques continue to be important: the observation of reaction rates continues to illuminate the manner in which enzymes act and how they may be inhibited. Some reactions generate striking spatial and sometimes chaotic patterns as they proceed (as on the pelts of animals, but in test-tubes too), and it is a challenge to physical chemists to account for the patterns by establishing their complex mechanisms and computing their complex and sometimes strikingly beautiful outcomes.