Medical Microbiology
Section 3 The conflicts
14 Immune defences in action
Introduction
The barrier effects of the skin and mucous membranes and their adjuncts such as cilia have already been referred to (see Ch. 9). We now turn to the back-up mechanisms called rapidly into play when an organism has penetrated these barriers – namely, complement, the phagocytic and cytotoxic cells, and a variety of cytotoxic molecules. While they lack the dramatic specificity and memory of adaptive (i.e. lymphocyte-based) immune mechanisms, these natural defences are vital to survival – particularly in invertebrates, where they are the only defence against infection (adaptive responses only evolved with the earliest vertebrates).
In addition to these non-specific mechanisms, the immune system enables the specific recognition of antigens by T and B cells as part of adaptive immunity. Broadly speaking, antibodies are particularly important in combating infection by extracellular microbes, particularly pyogenic bacteria, while T-cell immunity is required to control intracellular infections with bacteria, viruses, fungi or protozoa. Their value is illustrated by the generally disastrous results of defects in T and/or B cells, or their products, discussed in more detail in Chapter 30. This chapter gives examples of how these different types of immunity contribute to the body’s defences against microbes.
Antimicrobial peptides protect the skin against invading bacteria
A number of proteins that are expressed at epithelial surfaces, and by polymorphonuclear leukocytes (PMNs), can have a direct antibacterial effect. These include β-defensins, dermicidins and cathelicidins. Defensins form 30–50% of neutrophil granules, and disrupt the lipid membranes of bacteria. Dermicidin is made by sweat glands and secreted into sweat; it is active against E. coli, Staphylococcus aureus and Candida albicans. The precursor cathelicidin protein is cleaved into two peptides, one of which, LL37, is not only toxic to microorganisms but also binds LPS. Mice whose PMNs and keratinocytes are unable to make cathelicidin become susceptible to infection with group A streptococcus. Cathelicidin also plays a role in immunity to M. tuberculosis, through its action on vitamin D.
An interesting innate defence mechanism is the formation of neutrophil extracellular traps or NETS. Neutrophil serine proteinases such as cathepsin G, neutrophil elastase and proteinase 3 can be exocytosed by neutrophils to form neutrophil extracellular traps with chromatin that bind both Gram-positive and Gram-negative bacteria (Fig. 14.1) Of course, the bacteria can fight back, in this case through secreting DNAases or by having capsules to prevent entrapment.
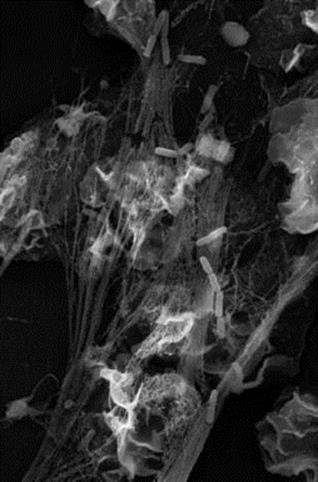
Figure 14.1 Neutrophil extracellular traps can trap bacteria. These chromatin-containing complexes can trap bacteria such as Shigella (illustrated).
(Photograph courtesy of Dr. Volker Brinkmann, Max Planck Institute for Infection Biology, Berlin.)
Lysozyme is one of the most abundant antimicrobial proteins in the lung. Genetically engineered transgenic mice that had a lot more lysozyme activity than control mice in their bronchoalveolar lavage were much better at killing group B streptococci, and Pseudomonas aeruginosa (Fig. 14.2).
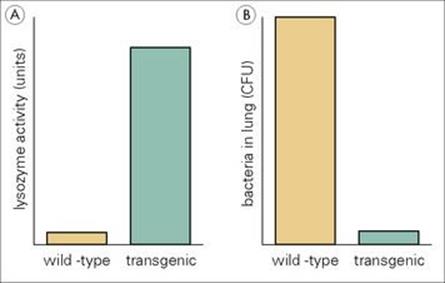
Figure 14.2 Transgenic mice making greater amounts of lysozyme are more resistant to infection with Pseudomonas aeruginosa. (A) The transgenic mice made 18-fold more lysozyme than the wild-type control mice. (B) The transgenic mice showed much greater killing of Pseudomonas aeruginosa in the lungs following intratracheal infection than the wild-type mice.
(Redrawn with data from Akinbi, H.T. et al. (2000) Bacterial killing is enhanced by expression of lysozyme in the lungs of transgenic mice. J Immunol 165:5760–5766.)
Complement
The alternative pathway and lectin binding pathways of complement activation are part of the early defence system
The basic biology of the complement system and its role in inducing the inflammatory response and promoting chemotaxis, phagocytosis and vascular permeability have been described in Chapter 9. Here we are concerned with its ability to directly damage microorganisms as part of the early response to infection. Contrary to what might be expected from the dramatic lysis of many kinds of bacteria in the test tube, the action of complement in vivo is restricted mainly to the Neisseria. Patients deficient in C5, C6, C7, C8 or C9 are unable to eliminate gonococci and meningococci, with the increased risk of developing septicaemia or becoming a carrier.
It should be emphasized that only the alternative pathway of complement activation or the mannan-binding lectin pathway form part of this natural ‘early defence’ system. Activation through the classical pathway occurs only after an antibody response has been made. It is not surprising to learn, therefore, that the alternative pathway appears to have evolved first.
Acute phase proteins and pattern recognition receptors
C-reactive protein is an antibacterial agent produced by liver cells in response to cytokines
Among the acute phase proteins produced in the course of most inflammatory reactions, C-reactive protein (CRP) is particularly interesting in being an antibacterial agent, albeit of very restricted range. CRP is a pentameric β-globulin, somewhat resembling a miniature version of IgM (molecular weight 130 000 compared with 900 000 for IgM). It reacts with phosphorylcholine in the wall of some streptococci and subsequently activates both complement and phagocytosis. CRP is produced by liver cells in response to cytokines, particularly interleukin-6 (IL-6, see Ch. 11), and levels can rise as much as 1000-fold in 24 h – a much more rapid response than that of antibody (see Ch. 9). Therefore, CRP levels are often used to monitor inflammation, for example, in rheumatic diseases. Most of the other acute phase proteins are produced in increased amounts early in infection and have not just antimicrobial activity but can act as opsonins, antiproteases, play an immunomodulatory role or be involved in the fibrinolytic or anticoagulant pathways. For example, many of the complement components are acute phase proteins. Those with a role in protection against infection are also termed pattern recognition receptors, such as mannose-binding lectin. Some acute phase proteins such as lipopolysaccharide binding protein may reduce pathology by binding toxic bacterial products such as lipopolysaccharide.
Macrophages can recognize bacteria as foreign using Toll-like receptors
Another family of surface receptors, called Toll-like receptors, on macrophages and other cells, bind conserved microbial molecules such as lipopolysaccharide (LPS) (endotoxin), bacterial DNA, double-stranded RNA or bacterial flagellin. Pattern recognition receptors recognize these repeated structures (pathogen-associated molecular patterns, see Ch. 9) and this leads to release of proinflammatory cytokines such as tumour necrosis factor alpha (TNFα), IL-1 and IL-6. Signalling through the Toll-like receptors also leads to the increased expression of major histocompatibility complex (MHC) molecules and of co-stimulatory molecules, thus enhancing antigen presentation and usually leading to the activation of T-helper 1 (Th1) cells. It was recently suggested that a number of rare single nucleotide polymorphisms within the TLR4 gene (TLR4 binds endotoxin) were more common in people with meningococcal disease compared with controls.
Microbes in the cytosol of a cell can also be recognized as foreign, using another family of pattern recognition receptors, called nucleotide-binding and oligomerization leucine-rich repeat receptors (NLR). Some NLRs can sense bacterial or viral DNA, leading to activation of inflammasomes, which are complexes of proteins, ultimately leading to the secretion of IL-1β and other proinflammatory cytokines. NLRs can also induce a process called autophagy, in which normal cytoplasmic contents are degraded after fusion with autolysosmes.
Collectins and ficolins
Collectins are proteins that bind to carbohydrate molecules expressed on bacterial and viral surfaces. This results in cell recruitment, activation of the alternative complement cascade, and macrophage activation. Two collectins, the surfactant proteins A and D, are able to directly inhibit bacterial growth and opsonize bacteria, leading to phagocytosis and activation of complement. Surfactant protein A, has been shown to play a role in the innate defence of the lung against infection with group B streptococci. Mice deficient in surfactant protein A were much more susceptible to infection, developing greater pulmonary infiltration and dissemination of bacteria to the spleen, compared with those able to produce the collectin. Polymorphisms in the surfactant A and D genes have also been linked to susceptibility to respiratory syncytial virus (RSV), as these surfactants act as opsonins for the virus.
Mannose-binding lectin (MBL) is another collectin found in serum. Binding of MBL to carbohydrates containing mannose on microorganisms leads to complement activation, through the mannan-binding lectin pathway. Bacteria opsonized by MBL bind to the C1q receptor on macrophages, leading to phagocytosis. Many individuals have low serum concentrations of MBL due to mutations in the MBL gene or its promoter. A recent study of children with malignancies showed that MBL deficiency increased the duration of infections. Lung surfactant proteins A and D, and MBL, bind to the surface spikes or S protein of the SARS virus (see Ch. 19), and so people with low MBL genotypes may be at increased risk of SARS infection.
Ficolins are plasma proteins with a similar structure to collectins, and bind N-acetyl glucosamine and lipotechoic acid from the cell walls of Gram-positive bacteria.
Fever
A raised temperature almost invariably accompanies infection (see Ch. 29). In many cases, the cause can be traced to the release of cytokines such as IL-1 or IL-6, which play important roles in both immunity and pathology (see Ch. 11). However, the interesting question as to whether the raised temperature itself is useful to the host remains unresolved.
It is probably unwise to generalize about the benefit or otherwise of fever
Several microorganisms have been shown to be susceptible to high temperature. This was the basis for the ‘fever therapy’ of syphilis by deliberate infection with blood-stage malaria, and the malaria parasite itself may also be damaged by high temperatures, though it is obviously not totally eliminated. In general, however, one would predict that successful parasites were those that were adapted to survive episodes of fever; indeed the ‘stress’ or ‘heat-shock’ proteins produced by both mammalian and microbial cells in response to stress of many kinds, including heat, are thought to be part of their protective strategy. On the other hand, several host immune mechanisms might also be expected to be more active at slightly higher temperatures: examples are complement activation, membrane function, lymphocyte proliferation and the synthesis of proteins such as antibody and cytokines.
Natural killer cells
Natural killer cells are a rapid but non-specific means of controlling viral and other intracellular infections
Natural killer (NK) cells provide an early source of cytokines and chemokines during infection, until there is time for the activation and expansion of antigen-specific T cells. NK cells can provide an important source of interferon-gamma (IFNγ) during the first few days of infection. NK cell cytokine production can be induced by monokines such as IL-12 and IL-18 that are in turn induced by macrophages in response to LPS or other microbial components. As well as IFNγ, NK cells can make TNFα and, under some conditions, the down-regulatory cytokine IL-10. Some tissues like the gut need their own special populations of NK-like cells. NK-like cells express some but not all the usual NK cell markers – but they do make large amounts of the cytokine IL-22, which helps defend the gut against certain intestinal pathogens.
NK cells can also act as cytotoxic effector cells, lysing host cells infected with viruses and some bacteria, as they make both cytotoxic granules and perforin. They recognize their targets by means of a series of activating and inhibitory receptors that are not antigen-specific. The inhibitory receptors recognize the complex of MHC class I and self peptide; if both this inhibitory receptor and another NK-cell-activating receptor are engaged, the NK cell will not be activated. However, if there is insufficient MHC class I on the cell surface, the inhibitory receptor is not engaged and the NK cell is activated to kill the target cell. This is an effective strategy, as some viruses inhibit MHC class I expression on the cells they infect. NK cells are therefore a more rapid but less specific means of controlling viral and other intracellular infections. The importance of NK cells is highlighted by the ability of mice lacking both T and B cells (severe combined immunodeficiency, SCID) to control some virus infections, and humans with NK cell defects are also susceptible to certain viruses (Table 14.1).
Table 14.1 Natural killer cells play an important role in controlling infections
|
Infections where NK cells have been shown to help control infection |
|
|
Human |
Mouse |
|
Human cytomegalovirus (HCMV; human herpesvirus 5) |
Mouse cytomegalovirus (MCMV) |
|
Vesicular stomatitits virus (VSV) |
Herpes simplex virus |
|
Herpes simplex virus (HSV) |
Vaccinia virus |
|
Human papilloma virus (HPV) |
Influenza virus |
NK cells form a bridge between the innate and adaptive immune responses, and their function may be enhanced by components of adaptive immunity. Some recent work even suggests that some NK cells can show some immunological memory, so perhaps their full functions are not yet fully appreciated!
Phagocytosis
Phagocytes engulf, kill and digest would-be parasites
Perhaps the greatest danger to the would-be parasite is to be recognized by a phagocytic cell, engulfed, killed and digested (Fig. 14.3). A description of the various stages of phagocytosis is given in Chapter 9. Phagocytes (principally macrophages) are normally found in the tissues where invading microorganisms are more likely to be encountered. In addition, phagocytes present in the blood (principally the PMNs) can be rapidly recruited into the tissues when and where required. Only about 1% of the normal adult bone marrow reserve of 3 × 1012 PMNs is present in the blood at any one time, representing a turnover of about 1011 PMNs/day. Most macrophages remain within the tissues, and well under 1% of our phagocytes are present in the blood as monocytes. PMNs are short lived, but macrophages can live for many years (see below).
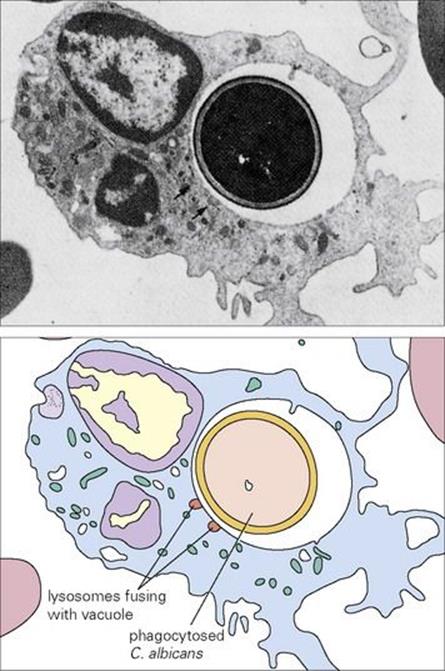
Figure 14.3 (A) Electron micrograph and (B) diagrammatic representation of neutrophil containing phagocytosed Candida albicans (× 7000).
(Courtesy of H. Valdimarsson.)
Intracellular killing by phagocytes
Phagocytes kill organisms using either an oxidative or a non-oxidative mechanism
The mechanisms by which phagocytes kill the organisms they ingest are traditionally divided into oxidative and non-oxidative, depending upon whether the cell consumes oxygen in the process. Respiration in PMNs is non-mitochondrial and anaerobic, and the burst of oxygen consumption, the so-called ‘respiratory burst’ (Fig. 14.4) that accompanies phagocytosis represents the generation of microbicidal reactive oxygen intermediates (ROIs).
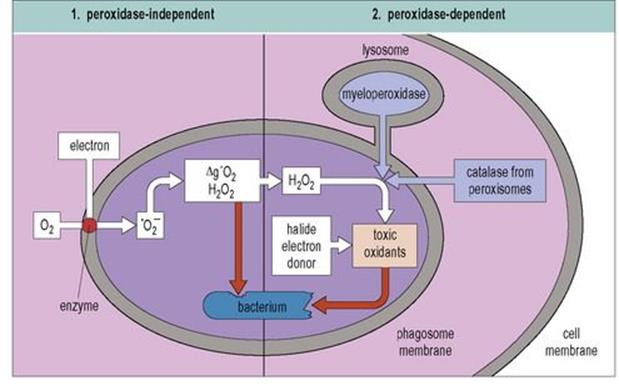
Figure 14.4 Oxygen-dependent microbicidal activity during the respiratory burst. The enzyme NADPH oxidase in the phagosome membrane reduces oxygen by the addition of electrons to form superoxide anion (![]() ). This can then give rise to hydroxyl radicals (•OH), singlet oxygen (Δg′O2) and H2O2, all of which are potentially toxic. If lysosome fusion occurs, myeloperoxidase or in some cases, catalase from peroxisomes, acts on peroxides in the presence of halides to generate toxic oxidants such as hypohalite. NADPH, nicotinamide adenine dinucleotide phosphate.
). This can then give rise to hydroxyl radicals (•OH), singlet oxygen (Δg′O2) and H2O2, all of which are potentially toxic. If lysosome fusion occurs, myeloperoxidase or in some cases, catalase from peroxisomes, acts on peroxides in the presence of halides to generate toxic oxidants such as hypohalite. NADPH, nicotinamide adenine dinucleotide phosphate.
(Reproduced from: Male, D., Brostoff, J., Roth, D.B., Roitt, I. (2006) Immunology, 7th edn. Mosby Elsevier, with permission.)
Oxidative killing
Oxidative killing involves the use of ROIs
The importance of ROIs in bacterial killing was revealed by the discovery that PMNs from patients with chronic granulomatous disease (CGD) did not consume oxygen after phagocytosing staphylococci. Patients with CGD have one of three kinds of genetic defect in a PMN membrane enzyme system involving nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, PHOX (see Ch. 30). The normal activity of this system is the progressive reduction of atmospheric oxygen to water with the production of ROIs such as the superoxide ion, hydrogen peroxide and free hydroxyl radicals, all of which can be extremely toxic to microorganisms (Table 14.2).
Table 14.2 Some organisms killed by reactive oxygen and nitrogen species
|
Bacteria |
Fungi |
Protozoa |
|
Staphylococcus aureus |
Candida albicans |
Plasmodium |
|
E. coli |
Aspergillus |
Leishmania (nitric oxide) |
|
Serratia marcescens |
CGD patients are unable to kill staphylococci and certain other bacteria and fungi, which consequently cause deep chronic abscesses. They can, however, deal with catalase-negative bacteria such as pneumococci because these produce, and do not destroy, their own hydrogen peroxide in sufficient amounts to interact with the cell myeloperoxidase, producing the highly toxic hypochlorous acid. The defective PMNs from CGD patients can be readily identified in vitro by their failure to reduce the yellow dye nitroblue tetrazolium to a blue compound (the ‘NBT test’, see Ch. 31).
The way in which ROIs actually kill microorganisms is controversial
ROIs can damage cell membranes (lipid peroxidation), DNA and proteins (including vital enzymes), but in some cases it may be the altered pH that accompanies the generation of ROIs that does the damage. Killing of some bacteria and fungi (e.g. E. coli, Candida) occurs only at an acid pH, while killing of others (e.g. staphylococci) occurs at an alkaline pH. There may also be a need for protease activity (e.g. cathepsins, elastase), with enzyme solubilization occurring as a result of the influx of H+ and K+ into the phagocytic vesicle.
Cytotoxic lipids prolong the activity of ROIs
As already mentioned, one of the targets of the toxic ROIs is lipid in cell membranes. ROIs are normally extremely short lived (fractions of a second), but their toxicity can be greatly prolonged by interaction with serum lipoproteins to form lipid peroxides. Lipid peroxides are stable for hours and can pass on the oxidative damage to cell membranes, both of the parasite (e.g. malaria-infected red cell) and of the host (e.g. vascular endothelium). The cytotoxic activity of normal human serum to some blood trypanosomes has been traced to the high-density lipoproteins, and in cotton rats to a macroglobulin.
Non-oxidative killing
Non-oxidative killing involves the use of the phagocyte’s cytotoxic granules
Oxygen is not always available for killing microorganisms; indeed, some bacteria grow best in anaerobic conditions (e.g. the Clostridia of gas gangrene), and oxygen would in any case be in short supply in a deep tissue abscess or a TB granuloma. Phagocytic cells therefore contain a number of other cytotoxic molecules. The best studied are the proteins in the various PMN granules (Table 14.3), which act on the contents of the phagosome as the granules fuse with it. Note that the transient fall in pH accompanying the respiratory burst enhances the activity of the cationic microbicidal proteins and defensins. Neutrophil serine proteinases have homology to the cytotoxic granzymes released by cytotoxic T cells.
Table 14.3 Contents of polymorphonuclear leukocyte (PMN) and eosinophil granules
|
PMN and eosinophil granule contents |
||
|
PMN |
Eosinophil |
|
|
Primary (azurophil) |
Specific (heterophil) |
Cationic |
|
Myeloperoxidase |
Lysozyme |
Peroxidase |
|
Acid hydrolases |
Lactoferrin |
Cationic proteins |
|
Cathepsins G, B, D |
Alkaline phosphatase |
ECP |
|
Defensins |
NADPH oxidase |
MBP |
|
BPI |
Collagenase |
Neurotoxin |
|
Cationic proteins |
Histaminase |
Lysophospholipase |
|
Lysozyme |
||
BPI, bactericidal permeability increasing protein; ECP, eosinophil cationic protein; MBP, major basic protein; NADPH, nicotinamide adenine dinucleotide phosphate.
Another phagocytic cell, the eosinophil, is particularly rich in cytotoxic granules (Table 14.3). The highly cationic (i.e. basic) contents of these granules give them their characteristic acidophilic staining pattern. Five distinct eosinophil cationic proteins are known and seem to be particularly toxic to parasitic worms, at least in vitro. Because of the enormous difference in size between parasitic worms and eosinophils, this type of damage is limited to the outer surfaces of the parasite. The eosinophilia typical of worm infections is presumably an attempt to cope with these large and almost indestructible parasites. Both the production and level of activity of eosinophils is regulated by T cells and macrophages and mediated by cytokines such as interleukin 5 (IL-5) and tumour necrosis factor alpha (TNFα).
Monocytes and macrophages also contain cytotoxic granules. Unlike PMNs (Table 14.4), macrophages contain little or no myeloperoxidase, but secrete large amounts of lysozyme. Lysozyme is an antibacterial molecule maintained at a concentration of about 30 mg/mL in serum, though this concentration can increase to as high as 800 mg/mL in rare cases of monocytic leukaemia. Macrophages are extremely sensitive to activation by bacterial products (e.g. LPS) and T-cell products, e.g. IFNγ. Activated macrophages have a greatly enhanced ability to kill both intracellular and extracellular targets.
Table 14.4 The major phagocytic cells – PMNs and macrophages – differ in a number of important respects
|
Polymorphonuclear leukocytes and macrophages compared |
||
|
PMN |
Macrophage |
|
|
Site of production |
Bone marrow |
Bone marrow |
|
Duration in marrow |
14 days |
54 h |
|
Duration in blood |
7–10 h |
20–40 h (monocyte) |
|
Average life span |
4 days |
Months–years |
|
Numbers in blood |
(2.5–7.5) × 109/L |
(0.2–0.8) × 109/L |
|
Marrow reserve |
10 × blood |
|
|
Numbers in tissues |
(Transient) |
100 × blood |
|
Principal killing mechanisms |
Oxidative, non-oxidative |
Oxidative, nitric oxide, cytokines |
|
Activated by |
TNFα, IFNγ, GM-CSF, microbial products |
TNFα, IFNγ, GM-CSF, microbial products (e.g. LPS) |
|
Important deficiencies |
CGD |
Lipid storage diseases |
|
Major secretory products |
Lysozyme |
Over 80, including: lysozyme, cytokines (TNFα, IL-1), complement factors |
CGD, chronic granulomatous disease; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; LPS, lipopolysaccharide; TNFα, tumour necrosis factor alpha.
Nitric oxide
A major secreted product of the activated macrophage is nitric oxide (NO), one of the reactive nitrogen intermediates (RNIs) generated during the conversion of arginine to citrulline by arginase. NO is strongly cytotoxic to a variety of cell types, and RNIs are generated in large amounts during infections (e.g. leishmaniasis, malaria). Arginase can also cause damage by leading to a deprivation of arginine, which is an essential amino acid for some viruses (e.g. herpes simplex) and parasites (e.g. the liver fluke Schistosoma).
Cytokines
Cytokines contribute to both infection control and infection pathology
Early studies with supernatants from cultures of lymphocytes and macrophages revealed a family of non-antigen-specific molecules with diverse activities, which were involved in cell-to-cell communication. These are now collectively known as ‘cytokines’. They play many crucial roles in protection against infectious diseases. The way in which these molecules acquired their sometimes rather misleading names, and the bewildering overlap of function between molecules of quite different structure, are described in detail in Chapter 11.
Cytokines are of importance in infectious disease for two contrasting reasons:
• They can contribute to the control of infection.
• They can contribute to the development of pathology.
The latter harmful aspect, of which TNFα in septic shock is a good example, is discussed in Chapter 17. The beneficial effects can be direct or more often indirect via the induction of some other antimicrobial process.
Interferons
The best-established antimicrobial cytokines are the interferons (IFNs) (Table 14.5). The name is derived from the demonstration in 1957 that virus-infected cells secreted a molecule that interfered with viral replication in bystander cells. IFNs of all three types (α, β and γ) interact with specific receptors on most cells, one for α and β and another for γ, following which they induce an antiviral state via the generation of at least two types of enzyme: a protein kinase and a 2′,5′-oligoadenylate synthetase. Both of these enzymes result in the inhibition of viral RNA translation and therefore of protein synthesis (Fig. 14.5).
Table 14.5 Human interferons (IFNs)
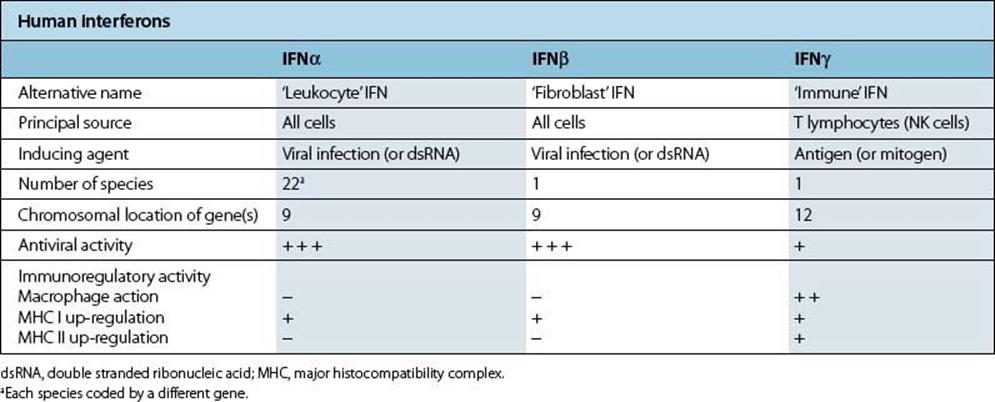
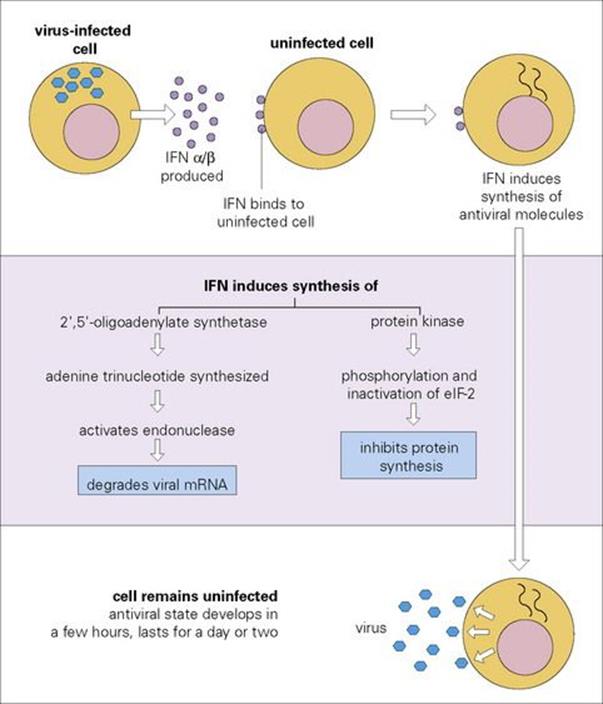
Figure 14.5 The molecular basis of type I interferon (IFN) action. eIF-2, eukaryotic initiation factor 2.
IFNα and IFNβ constitute a major part of the early response to viruses
IFNα and IFNβ (type I interferons) are produced rapidly within 24 h of infection, and constitute a major part of the early response to viruses.
Type I IFNs can also inhibit virus assembly at a later stage (e.g. retroviruses), while many of the other effects of IFN contribute to the antiviral state, for example, by the enhancement of cellular MHC expression and the activation of NK cells and macrophages (Fig. 14.6). Unlike cytotoxic T cells, IFN normally inhibits viruses without damaging the host cell. Although best known for their antiviral activity, type I IFNs have recently been shown to be induced by, and active against, infections with a wide range of organisms, including rickettsia, mycobacteria and several protozoa. A recent study of gene expression in patients with tuberculosis identified that many genes induced by type I interferons as well as by type II IFNγ were activated.
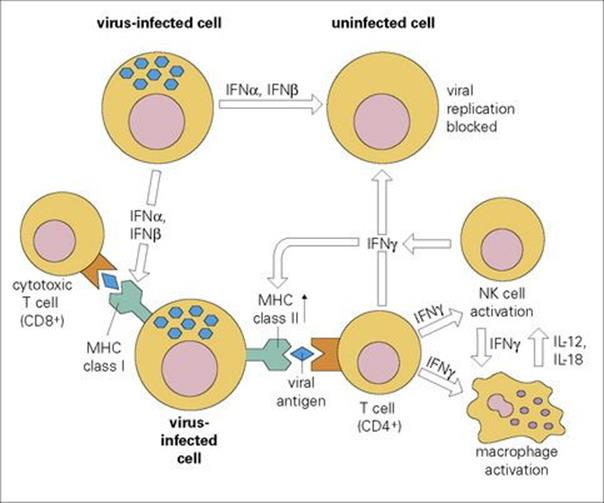
Figure 14.6 The multiple activities of interferons (IFNs) in viral immunity. MHC, major histocompatibility complex; NK, natural killer.
In animal experiments, treatment with antibodies to IFNα greatly increases susceptibility to viral infection; treatment with IFNα has proved useful for some human virus infections, notably chronic hepatitis B (see Ch. 22).
IFNγ (type II, immune interferon) is mainly a T-cell product and is therefore produced later, although, as discussed above, an early IFNγ response may be mounted by NK cells. The role of IFNγ is discussed further under T cells, below. Some intracellular organisms (e.g. Leishmania) can counteract the effect of IFNγ on MHC expression, thereby facilitating their own survival.
Other cytokines
TNFα production can be good or bad
A striking example of a potentially useful role for TNFα in infection is illustrated by what happened when a humanized antibody against tumour necrosis factor alpha was used to treat patients with rheumatoid arthritis and Crohn’s disease. A number of treated patients developed tuberculosis soon after starting therapy (Fig. 14.7); others developed Listeria, Pneumocystis or Aspergillus infections. Patients should now be tested for latent tuberculosis before starting treatment with the tumour necrosis blocking antibody. However, TNF is also thought to contribute to the pathology of tuberculosis, as well as that of malaria (see Ch. 12). This illustrates the often confusing role that cytokines play in infectious diseases of all kinds – ‘enough is enough’ and ‘too much is dangerous’ seem to be the rules for these powerful molecules. Paradoxically, TNF concentration is raised in HIV infection and has been found to enhance the replication of HIV in T cells – a ‘positive feedback’ with worrying potential. The role of T-cell-derived cytokines such as IFNγ in immunity to infection is discussed below.
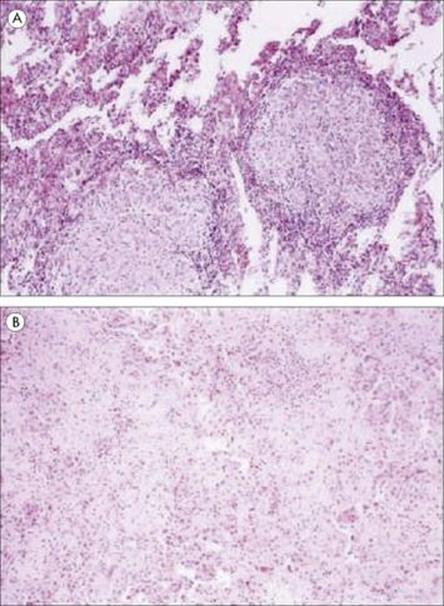
Figure 14.7 Photomicrographs of lung specimens from patients with tuberculosis (A) who did not (× 100) or (B) who did (× 100) receive Infliximab, a humanized antibody to TNFα. In the patient without Infliximab treatment, the well-formed granuloma shows little necrosis; in the patient with anti-TNF treatment, there is minimal granuloma formation but much fibrosis and inflammation.
(Reproduced from Keane, J. et al. (2001) Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med 345:1098–1104, with permission.)
Antibody-mediated immunity
The key property of the antibody molecule is to bind specifically to antigens on the foreign microbe. In many cases, this is followed by secondary binding to other cells or molecules of the immune system (e.g. phagocytes, complement). These are discussed below, but first some general features that influence the effectiveness of the antibody response should be mentioned.
Speed, amount and duration
Because of the cell interactions involved and the need for proliferation of a small number of specific precursor lymphocytes, a primary antibody response can be dangerously slow in reaching protective levels. The classic example, before penicillin, was lobar pneumonia, where the race between bacterial multiplication and antibody production was ‘neck-and-neck’ for about 1 week, at which point one side or the other dramatically won. Nowadays, of course, vaccines and antibiotics have intervened to improve the patient’s chances. Experiments with specially bred lines of mice suggest that the speed and size of an antibody response is under the control of a large number of genes, and the same is undoubtedly true in humans.
The rate of replication of the microorganism must also be considered. Replication rates, as indicated by doubling times (see Ch. 15) vary from < 1 h (most viruses, many bacteria) to days or even weeks (mycobacteria, T. pallidum). Microorganisms tend to grow more slowly in vivo than in vitro, which shows that the host environment is generally hostile. When the incubation period is only a few days (e.g. rhinovirus, rotavirus, cholera) the antibody response is too slow to affect the initial outcome, and rapidly produced cytokines such as interferons are more important.
Generally speaking, the antibody response continues as long as antigen is present, although some down-regulation may occur in very prolonged responses, presumably in an effort to limit immunopathology (see Ch. 17). The lifelong immunity that follows many virus infections may often be due to regular boosting by viruses in the community, but sometimes (e.g. yellow fever) there is no obvious boost yet antibodies persist for decades. Such persistence of immunological memory may be due to the non-specific stimulation of memory B and T cells by cytokines during responses to other antigens, a process called bystander activation.
Affinity
It seems self-evident that a higher antigen-binding affinity would render antibody more useful, and passive protection experiments have confirmed this. Affinity is determined by both the germline antibody gene pool and somatic mutation in individual B lymphocytes, and appears to be under genetic control that is separate from that controlling the total amount of antibody made. A tendency towards a low antibody affinity to the tetanus toxoid vaccine has been found in some subjects, particularly those with predominantly IgG4 responses, and there is strong evidence from mouse experiments that failure to develop high-affinity antibody responses can predispose to immune complex disease.
Antibody classes and subclasses (isotypes)
The different Fc portions of the antibody molecule are responsible for most of the differences in antibody function (see Ch. 10). Switching from one to another while preserving the same Fab portion allows the immune system to ‘try out’ different effector mechanisms against the microbial invader. This flexibility is not total. For example, T-independent antigens such as some polysaccharides induce only IgM antibodies, T cells being required for the switch to IgG, IgA or IgE. IgG antipolysaccharide responses tend to be mainly IgG2, whereas antiprotein IgG is mainly IgG1. The poor development of IgG2 in children below the age of about 2 years explains their lack of response to bacteria with polysaccharide capsules (e.g. Strep. pneumoniae, Haemophilus influenzae), and considerable effort is being made to produce vaccines that would induce other subclasses of IgG. Antibodies to viruses are predominantly IgG1 and IgG3, and to helminths, IgG4 and IgE, while antigens encountered via the digestive tract induce mainly IgA, the only type of antibody that can function in the protease-rich intestinal environment; T cells and cytokines also play important roles in these isotype preferences.
Blocking and neutralizing effects of antibody
Simple binding of antibody molecules to a microbial surface is often enough to protect the host. It may physically interfere with the receptor interaction necessary for microbial entry (e.g. of a virus into a cell) or with the binding of a toxin to its host receptor. This is the basis of many life-saving vaccines against viruses or bacterial toxins.
Blocking of attachment and entry can be effective against all organisms that use specific attachment sites, whether viral, bacterial or protozoal (see Ch. 16). An important exception is those organisms that parasitize the macrophage, such as the virus of dengue fever; here the presence of a low concentration of IgG antibody can actually enhance infection by promoting attachment to Fc receptors (see below).
A more subtle blocking effect of antibody is interference with essential surface components of the parasite, particularly if these are enzymes or transport molecules. Needless to say, the successful parasite takes steps to protect such components whenever possible, as described in Chapter 16.
Immobilization and agglutination
Immunoglobulin antibodies, particularly the large, pentameric IgM, are the same order of size as some of the smaller viruses, and larger than the thickness of a bacterial flagellum (Fig. 14.8), so the simple physical attachment of antibody can considerably restrict the activities of motile organisms. In addition, the multivalent design of the antibody molecules enables it to link together two or more organisms, as can readily be demonstrated in the bacterial agglutination tests (Fig. 14.9). The protective value of agglutination in vivo is hard to assess; once clumped, most organisms are probably rapidly phagocytosed, but clumps of still motile trypanosomes can be seen in the blood of infected animals with enough serum antibodies. Agglutination reactions in vitro are very useful in diagnosis (see Ch. 31).
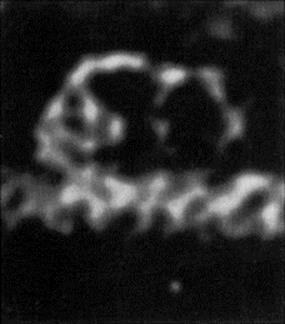
Figure 14.8 Electron micrograph of an IgM molecule. The crab-like configuration is due to cross-linkage with a single flagellum. As an independent molecule, it adopts a ‘star-line’ configuration.
(Courtesy of A. Feinstein.)
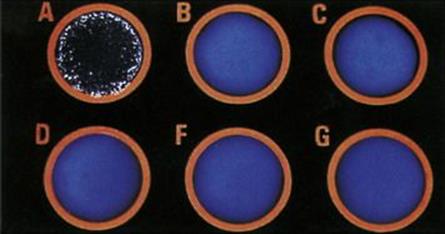
Figure 14.9 Bacterial agglutination. Agglutination of group A streptococcus with latex particles coated with anti-group A antibodies.
(Courtesy of D.K. Banerjee.)
Lysis
Lysis of bacteria in the presence of complement provides another convenient assay for the presence of antibody. However, lysis probably plays a major protective role in only a restricted range of infections, notably those caused by Neisseria and some viruses (see Ch. 17).
Opsonization
Whether by the direct binding of the immunoglobulin CH2 and CH3 regions to Fc receptors, or via the activation of complement to allow C3b to bind to its receptor, opsonization represents the most important overall function of the antibody molecule. Telling evidence for this is the general similarity in the effects on the patient of defects in antibody, complement (up to and including C3) and phagocytic cells (see Ch. 30). It is estimated that the rate of phagocytosis is enhanced by up to 1000-fold by antibody and complement acting together (Fig. 14.10). Lobar pneumonia due toStrep. pneumoniae again provides a good example: IgG antibody against the capsule allows neutrophils to phagocytose the organisms, converting overnight a lung virtually solid with fluid, fibrin and phagocytic cells into the normal breathing apparatus. Note that the later complement components C5–9 are not required, so that deficiencies of these do not predispose to bacterial infection in general (see Ch. 30). Of course, the effectiveness of opsonization depends on the phagocytic cell being capable of finishing off the ingested organism. This is not the case, however, with organisms that inhibit or avoid the normal intracellular killing processes, of which mycobacteria are a typical example (see Ch. 16).
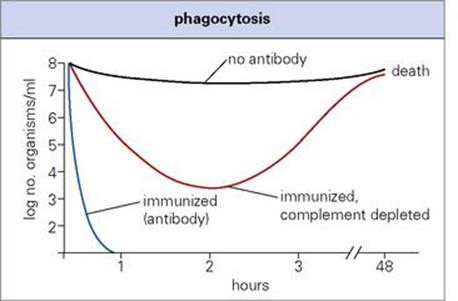
Figure 14.10 Phagocytosis. Antibody and complement accelerate the clearance of pneumococci from the blood of mice.
Antibody-dependent cellular cytotoxicity
In the case of larger organisms (worms being the most obvious example), phagocytosis is clearly not a possibility. However, several types of cell, having made contact with the parasite through antibody and Fc receptors in the same way as phagocytes do, can inflict damage extracellularly. These include most conventional phagocytes as well as eosinophils and platelets. It must be said that virtually all the evidence for this kind of mechanism comes from experiments done in vitro, and here again it is extremely difficult to assess their role in vivo.
Indeed, the precise way in which antibody protects against infection is, in the majority of cases, still unknown. For example, the enormous production of IgA in the intestine, which may amount to half of all antibody produced in the body, suggests the vital importance of mucosal protection, and yet deficiency of IgA is relatively common and not particularly serious.
Table 14.6 gives some examples of common infections normally controlled by antibody. Once again, it must be emphasized that the presence of antibody by no means denotes a protective role. It may be directed against irrelevant or non-critical microbial antigens, or the infection may be of a type that is not controlled by antibody, as with many intracellular infections (tuberculosis, typhoid, herpes virus). The best indication of the value of antibody comes from antibody-deficiency syndromes (Ch. 30).
Table 14.6 Antibody and cell-mediated immunity (CMI) in resistance to systemic infections
|
Antibody and CMI in resistance to systemic infections |
||
|
Type of resistance |
Antibody |
CMI |
|
Recovery from primary infection |
Yellow fever, polioviruses, coxsackieviruses |
Poxviruses: e.g. ectromelia (mice), vaccinia (humans) |
|
Resistance to re-infection |
Nearly all viruses including measles, most bacteria |
Tuberculosis |
|
Resistance to reactivation of latent infection |
Varicella-zoster, cytomegalovirus, Herpes simplex, tuberculosis, Pneumocystis jirovecic |
|
Either antibody or CMI is known to be the major factor in these examples. But in many other infections there is no information, and sometimes both types of immunity are important. LCM lymphocytic choriomeningitis.
a Protection is incomplete and short-lived.
b Both Th1 and Th17 cells may be involved
c Formerly P. carinii.
Cell-mediated immunity
T cells form the second main component of the adaptive immune response (Chs 10 and 11). Some act by producing cytokines that induce macrophage activation or help antibody production, others by their direct cytotoxic action on infected target cells. In both cases, the T cell needs to ‘see’ the combination of specific peptide and MHC molecule that is recognized by its T-cell receptor. Some examples of the importance of antibody and cell-mediated immunity in resistance to systemic infections are given in Table 14.6.
T-cell immunity correlates with control of bacterial growth in leprosy
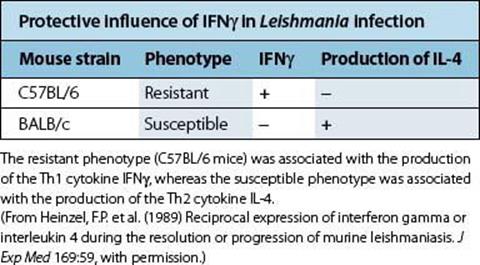
In leprosy, there is a spectrum of disease, ranging from the paucibacillary tuberculoid form to the multibacillary lepromatous disease. M. leprae-specific T-cell immunity, as measured by lymphocyte proliferation, secretion of Th1 cytokines such as IFNγ, or delayed-type hypersensitivity skin testing, is found in patients with tuberculoid leprosy, but absent in patients with lepromatous leprosy. The value of T-cell stimulation leading to macrophage activation and bacterial killing is clearly illustrated by experiments in which lepromatous leprosy patients’ skin lesions were injected with IFNγ. This resulted in an influx of T cells and macrophages into the skin lesions, and a reduction in the number of bacteria. Another good example of the protective role of IFNγ and Th1 immunity is seen in animal models of Leishmania infection: i.e. some mouse strains such as C57BL/6 are resistant to disease, controlling the infection and making a good Th1 cytokine response, whereas other susceptible strains such as BALB/c cannot control parasite growth and fail to make IFNγ (Table 14.7).
Table 14.7 Cytokine production in the spleens of mice infected with Leishmania major
Further evidence for the protective effects of IFNγ
The protective effects of making IFNγ, which then binds to its specific receptor on macrophages and induces macrophage activation and the production of antimicrobial molecules, are illustrated very clearly by the consequences of a failure in IFNγ synthesis or of binding to its receptor. Mice in which the gene for IFNγ has been inactivated (‘knocked-out’) become very susceptible to intracellular infections. Rare individuals with mutations in the genes for the IFNγ receptor have been identified. Such individuals are susceptible to infections with mycobacteria, or to disseminated infections following BCG vaccination (Fig. 14.11).
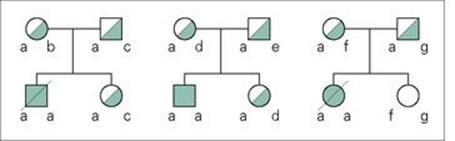
Figure 14.11 Genetic mutations in the IFNγ receptor cause susceptibility to mycobacterial infections. Three Maltese families had children who were susceptible to atypical mycobacterial infection (solid symbols), two of whom died (slashed symbols). Individuals with carrier status are shown with half-filled symbols. All of the affected children were homozygous for the disease locus a on chromosome 6q22-q23, with a point mutation in the gene for the IFNγ receptor. This mutation introduces a stop codon resulting in a non-functional truncated protein.
(From: Newport, M.J. et al. (1996) A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N Engl J Med 335:1941–1949, with permission. © Massachusetts Medical Society.)
Some bacteria evade protective Th1 responses by inducing antigen-specific regulatory T cells. Bordetella pertussis infection induces regulatory T cells specific for its filamentous haemagglutinin and pertactin. These regulatory T cells produce IL-10 which then suppresses Th1 immunity.
Human IFNγ and IFNα are now used to treat a number of infections (Table 14.8).
Table 14.8 Examples of the therapeutic use of cytokines in infectious diseases in humans
|
Cytokine |
Organisms |
Comments |
|
IFNα |
Hepatitis B, Hepatitis C |
Pegylated formulation; for HCV usually given with ribavirin/protease inhibitors |
|
Human herpesvirus 8 |
Intralesional injection of Kaposi’s sarcoma plaques |
|
|
Human papillomavirus |
Intralesional therapy for genital lesions |
|
|
IFNγ |
Hepatitis B |
Pegylated formulation |
|
M. tuberculosis |
Aerosolized treatment given on trial basis to patients with drug-resistant TB and atypical mycobacteria |
|
|
Cryptococcus neoformans |
Combined with standard antifungal therapy in a phase II trial with a trend towards improved outcomes |
|
|
General |
Given to patients with chronic granulomatous disease to prevent bacterial and fungal infections |
The evidence of benefit for some of these treatments is stronger than for others. The main clinical use of a cytokine is the treatment of patients with chronic granulomatous disease with IFNγ to prevent bacterial and fungal infections.
Cytokine signatures
During viral infections, the pattern or signature of cytokines produced by T cells may vary with clearance of infection, or antigen load during a chronic infection (Fig. 14.12). For example, primary infection with HIV or CMV induces mainly IFNγ-producing T cells; in influenza IL-2-producing cells predominate after viral clearance; chronic viral infection such as EBV or HIV in non-progressors seem to lead to a mixed IFNγ and IL-2 signature, but with progressive HIV infection and a higher antigen load this shifts to dominant IFNγ production. The balance between effector T cells and resting memory T cells will also change from acute to chronic disease with HIV. Healthy people have balanced populations of naive, effector and memory T cells in both the CD4 and CD8 compartments; in acute HIV, the effector CD8 T cells expand, but with chronic infection the naive and memory CD4 T cells are lost.
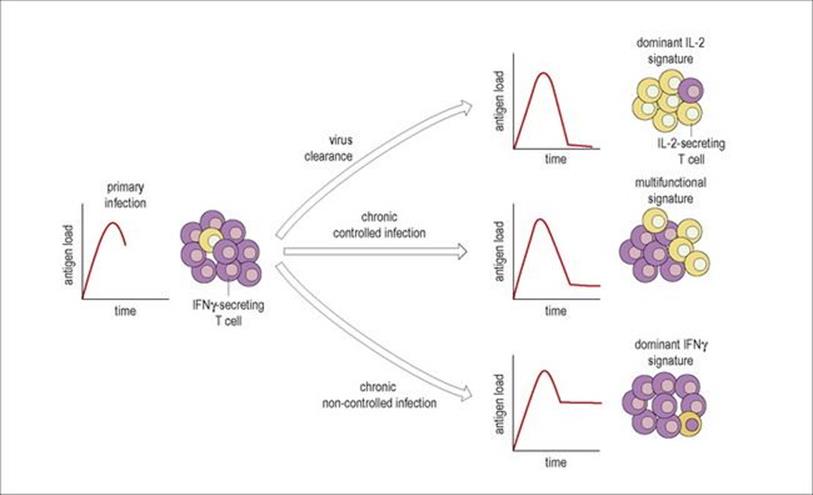
Figure 14.12 Cytokine signatures. The balance between virus-specific T cells secreting IFNγ (purple cells) and T cells secreting IL-2 (yellow cells) is shown for different types of viral infection, illustrated by the small graphs of antigen load over time. Acute infections such as primary HIV-1 or CMV have a high antigen load; following clearance of virus with an infection such as influenza the antigen load falls and IL-2-producing cells predominate; in a chronic controlled infection such as EBV, chronic CMV or HIV-1 in long-term non-progressors there is production of both IL-2 and IFNγ-secreting T cells, and in chronic infection with high antigen load such as progressive HIV-1 infection, IFNγ-producing cells predominate.
(Redrawn from: Pantaleo, G., Harari, A. (2006) Functional signatures in antiviral T-cell immunity for monitoring virus-associated diseases. Nat Rev Immunol 6:417–423.)
Th17 T cells
The division of CD4 T cells into Th1 and Th2 T cells has aided our understanding of immunity to many infections. However, another CD4 subset making IL-17, and so called Th17, and induced by the cytokine IL-23, contributes to antimicrobial immunity. Th17 cells play a role in immunity against a number of bacterial infections including Klebsiella pneumoniae, E. coli, Staph. aureus, Listeria monocytogenes and Candida albicans. One way in which IL-17 works is by inducing neutrophil recruitment. Some patients with chronic mucocutaneous candidiasis have signalling defects leading to problems with production of Th17 cells and increased susceptibility to Candida. IL-17 along with IL-22 may also help restrict tissue damage during episodes of inflammation.
Is a positive delayed-type hypersensitivity skin test an indicator of immunity?
The most widely used test of T-cell immunity in humans is the delayed-type hypersensitivity (DTH) skin test, in which induration induced by the intradermal injection of antigen is measured 2–3 days later. Such tests can be used to screen for T-cell anergy, e.g. by using candidin, as most individuals will have been exposed to Candida. The most widely used skin test is the Mantoux skin test using antigens from M. tuberculosis. However, this test is neither diagnostic nor a correlate of immunity. Unfortunately, many of the antigens in the purified protein derivative of M. tuberculosis used as the antigen in this test are cross-reactive with those in other mycobacteria, including BCG and non-tuberculous environmental mycobacteria. This means that skin test positivity may be found in BCG-vaccinated subjects, and in people not exposed to M. tuberculosisitself. Nevertheless, those with a large skin test response are at increased risk of developing tuberculosis, showing that strong T-cell responses can be induced during disease progression and that a strongly positive skin test can indicate infection rather than immunity. More specific interferon-gamma release assays that measure IFNγ release in response to antigens present in M. tuberculosis and not found in BCG or most environmental mycobacteria are now available.
Cytotoxic T lymphocytes kill by inducing ‘leaks’ in the target cell
The well-known cytotoxic T lymphocyte (CTL) is unusual in that both antigen-specific recognition and killing of the target are carried out by the same cell. The recognition step, involving an antigenic fragment that becomes associated with a class I MHC molecule, is discussed in Chapter 10, and displays the high degree of specificity characteristic of adaptive responses. The killing mechanism, however, is relatively non-specific. It appears to involve the induction of ‘leaks’ or pores in the target cell membrane by the insertion of perforin, a 66 kDa molecule that is structurally and functionally similar to the terminal complement component C9 (80 kDa; Fig. 14.13). Other molecules, including granzymes and cytokines such as TNFα, may also be involved, and their effects may be either direct or indirect. Target cell death may be due to:
• leakage
• induction of apoptosis: a ‘suicide’ programme built into all cells and induced by Fas/FasL interactions, granzymes and TNFα.
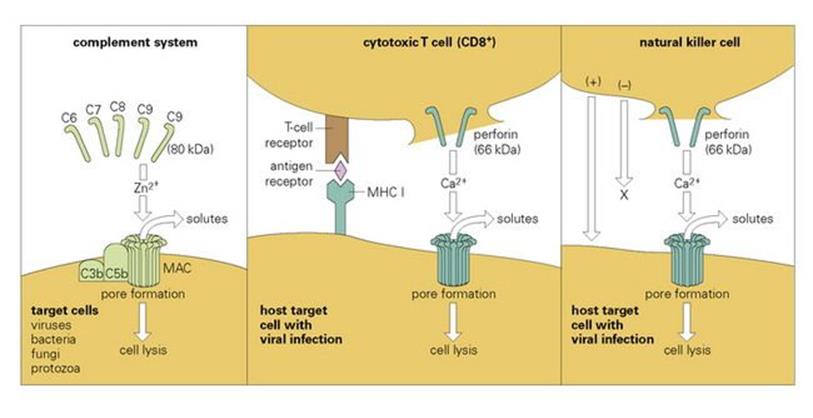
Figure 14.13 Comparison of the lytic mechanisms of cytotoxic cells and the complement system. Ca2 +, calcium; MAC, membrane attack complex; MHC, major histocompatability complex; Zn2 +, zinc.
These mechanisms are thought to operate principally against virus-infected cells, but some cells infected with other intracellular parasites, including mycobacteria (e.g. M. leprae in Schwann cells) and even protozoa (e.g. Theileria parva in lymphocytes) may also be susceptible. Most cytotoxic T cells are CD8-positive, recognizing MHC class- I-restricted peptide epitopes, but cytotoxicity can also be mediated by CD4 T cells and by γδ T cells. CD8 T cells are activated in some bacterial infections such as tuberculosis, where the microbe or its antigens may escape into the cytoplasm. CD8 activation may also result from a process called cross-priming, where bacterial antigens taken up by a dendritic cell are processed not only for MHC class II but also for MHC class I presentation. In some cases, apoptotic blebs released by apoptotic infected macrophages may be taken up by the dendritic cells. The lysis of an infected target cell may not always kill the intracellular microbe, but its release from its hideaway may lead to phagocytosis and subsequent killing by a more highly activated macrophage (Fig. 14.14).
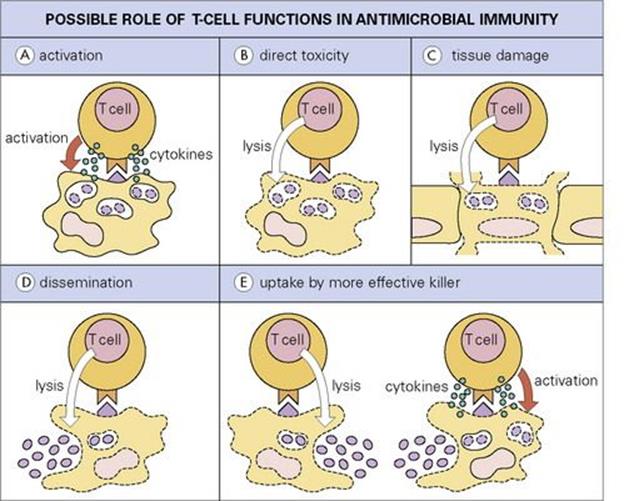
Figure 14.14 Possible roles for T cells in immunity to intracellular microbes. (A) The T cell activates intracellular killing mechanisms by secretion of cytokines such as IFNγ, e.g. in a macrophage. (B) The T cell directly kills cell and parasite. (C) The T cell destroys vital tissue in the process of killing the parasite. (D) By lysing cells the T cell allows still-living parasites to disseminate. (E) Parasites released in this way may be phagocytosed by a more effective host cell.
(Redrawn from: Kaufmann, S.H. (1989) In vitro analysis of the cellular mechanisms involved in immunity to tuberculosis. Rev Infect Dis 11(Suppl 2):S448–S454.)
Another interesting recent finding is that not all CD8 T cells can act as effector cytotoxic T cells. More human CD8 T cells express the granule protease granzyme A than the pre-formed effector molecule perforin. In HIV infection, two-thirds of the CD8 T cells express granzymes but only one-third express perforin. This may explain why virus-infected cells escape killing by antigen-specific CD8 T cells in HIV infection.
A summary of cytotoxic molecules made by cells involved in both natural and adaptive immunity is given in Table 14.9.
Table 14.9 Some important cytotoxic molecules that operate against infectious organisms
|
Cytotoxic molecules |
||
|
Host component |
||
|
Major cell source |
Molecule |
Effective against |
|
Liver cells |
Complement C1–3 |
Bacteria, fungi |
|
Macrophage |
Complement C5–9 |
Neisseria |
|
Macrophage, neutrophil, eosinophil |
Reactive oxygen intermediates (plus peroxidase) |
Bacteria, fungi, malaria |
|
Macrophage |
Lysozyme |
Gram-positive bacteria |
|
Interferon (α, β) |
Viruses |
|
|
Tumor necrosis factor |
Viruses, bacteria, malaria |
|
|
Reactive nitrogen intermediates |
Leishmania, malaria |
|
|
Neutrophil |
Defensins |
Bacteria, fungi |
|
Cathepsins |
Bacteria, fungi |
|
|
Lactoferrin |
Bacteria, yeasts |
|
|
Eosinophil |
Cationic proteins |
Schistosome worms |
|
T lymphocyte |
Cytokines |
|
|
Perforins |
||
|
Granzymes |
||
|
Granulysin |
Bacteria, fungi |
|
|
Natural killer cell |
Perforins |
|
|
Granzymes |
||
|
Granulysin |
Bacteria, fungi |
|
|
Liver, fat |
High density lipoprotein |
Trypanosomes |
|
Low density lipoprotein (oxidized) |
Malaria |
|
|
Kidney |
Urea |
Bacteria |
Recovery from infection
The everyday concept of an infectious disease is one where the patient is ill for a period of days to months and then recovers. In some cases, they are subsequently immune to the disease. In such circumstances, one can be fairly certain that adaptive (lymphocyte-based) mechanisms have been at work, since: (1) the existence of disease symptoms implies that natural defence mechanisms, which act rapidly, did not succeed in eliminating the parasite; (2) a period of days or weeks is typical of the time that adaptive immune mechanisms take to reach maximal levels; and (3) subsequent immunity is a sign of the immunological memory exclusive to lymphocytes, which possess the ability to specifically recognize antigens, to proliferate into clones, and to survive as memory cells. Thus, the older individuals are, the better they are adapted to the environment, until old age begins to weaken the immune system itself.
In the early stages of an infection, however, adaptive immunity can appear somewhat clumsy and ineffective. Since the lymphocytes are programmed to recognize the shapes of antigenic epitopes, they cannot distinguish virulent from harmless parasites, and must rely on recognizing “danger” signals – nor can they ‘know’ which type of immune response will be most effective. Often, one mechanism is responsible for recovery and another for resistance to re-infection (e.g. cytotoxic cells and interferon in recovery from measles, antibody in prevention of a second attack). In many infections, there is still controversy as to which of the numerous responses that can be detected are useful, harmful or neutral. The reason for an individual’s failure to recover from, or to suffer from, an infection can also be hard to pinpoint. If the infection is one from which most people recover (e.g. measles), or from which they do not suffer at all (e.g. Pneumocystis), an immunodeficiency should be considered (see Ch. 30). Infections that are rapidly fatal in normal individuals (e.g. Lassa fever) are frequently those to which the human immune system has not been exposed, since they are normally maintained in animals and only accidentally infect humans (see Zoonoses, Ch. 28). But if the infection normally runs a prolonged course without either being eliminated or killing the host, the parasite can be considered to be successful, and this success will be due to one or more survival strategies. These are the subject of Chapter 16.
Nutrition may have more subtle effects on immunity to infection
Even if an immunodeficiency state is not present, other factors may affect how a person copes with an infection. For example, during starvation or malnutrition, concentrations of the hormone leptin (which is produced by adipocytes and among other functions induces PMN activation) fall. Mice fasted for 2 days had higher numbers of Strep. pneumoniae in their lungs than normally fed animals, but if the fasted animals were given leptin, the number of PMNs in the lungs increased and the bacterial counts fell (Fig. 14.15). Leptin-deficient mice are highly susceptible to bacterial infections such as Klebsiella and Listeria. However, being obese is not good either – it is worth noting that obese people seem to be more susceptible to many more types of infections than those of a normal weight.
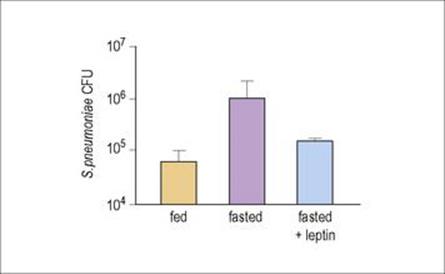
Figure 14.15 Leptin can restore host defence against Strep. pneumoniae in fasted mice. Colony-forming units (CFU) of bacteria in the lung were measured after normal feeding (orange column), in animals fasted for 48 h (purple column), or fasted but given leptin (blue column), 24 h after infection with Strep. pneumoniae.
(Redrawn from: Mancuso, P. et al. (2006) Leptin corrects host defense defects after acute starvation in murine pneumococcal pneumonia. Am J Respir Crit Care Med 173:212–218.)
![]()
 Key Facts
Key Facts
• Protection against infectious organisms that penetrate the outer barriers of the skin and mucous membranes is mediated by a variety of early defence mechanisms, which constitute innate immunity.
• These early defence mechanisms occur more rapidly but are less specific than the adaptive mechanisms based on lymphocyte (T and B cell) responses.
• Important early defence mechanisms include the acute phase response, the complement system, IFNs, phagocytic cells and NK cells. Together, these act as a first line of defence during the initial hours or days of infection.
• Adaptive immunity, mediated by antibody and T cells, is responsible for recovery from infection in many cases, although these mechanisms take days to weeks to reach peak efficiency.
• Sometimes, as in the common viral infections, cell-mediated immunity is responsible for recovery from infection, and antibody for the maintenance of immunity.
• Failure to recover from infection may be due to some deficiency of host immunity or to successful evasion strategies used by the microorganism.
![]()