Medical Microbiology
Section 3 The conflicts
17 Pathologic consequences of infection
Introduction
Symptoms of infections are produced by the microorganisms or by the host’s immune responses
Symptoms that appear rapidly after the acquisition of an infection are usually due to the direct action of the invading microbe by what it secretes. Thus a virus in a cell may cause metabolic ‘shut-down’ or lyse the cell. Bacteria, however, provoke most of their acute effects by releasing toxins, but may also cause distress by inducing inflammation. The inflammatory response is, of course, an important component of host protection, vascular permeability being vital for the rapid mobilization of cells such as neutrophils, and serum components such as complement and antibody. Inflammation is therefore intrinsically a healthy sign, and it is interesting that some virulent bacteria (e.g. staphylococci) can, to some extent, inhibit the inflammatory response.
Often, however, pathologic changes are secondary to the activation of immunologic mechanisms that are normally thought of as protective. These may involve innate or the adaptive immune system or, more usually, both (Fig. 17.1). Tissue damage resulting from adaptive immune responses is usually referred to as ‘immunopathology’ and is quite common in infectious diseases, particularly those that are chronic and persistent. The immunologic basis of these mechanisms of tissue damage is described in Chapter 11.
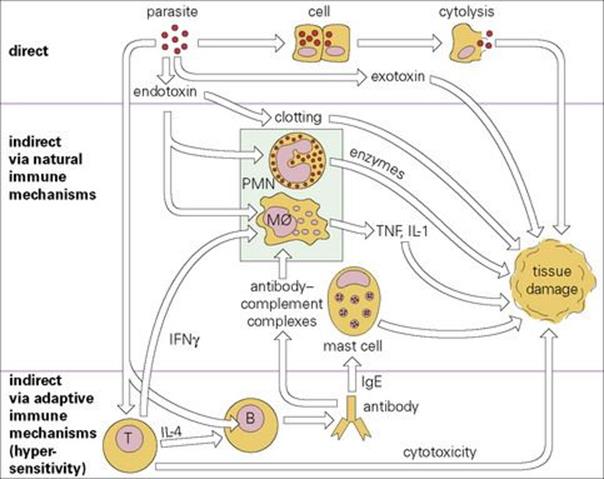
Figure 17.1 Pathologic effects of infection: a general scheme. Infectious parasitic organisms can cause disease directly (top) or indirectly via overactivation of various immune mechanisms, either natural (middle) or adaptive (bottom). IFN, interferon; IL, interleukin; Mφ, macrophage; PMN, polymorphonuclear leukocyte; TNF, tumour necrosis factor.
Certain viruses can cause permanent malignant change in cells as a result of direct, indirect and a mixture of both mechanisms. Seven viruses that infect humans cause up to 15% of human cancers around the world. These include HTLV type 1 (lymphomas, leukaemias), Epstein–Barr virus (nasopharyngeal carcinoma and Burkitt’s lymphoma), human papillomaviruses (cervical cancer), hepatitis B and C virus infections (liver cancer), HIV (immunosuppression leads to the development of cancers associated with KSHV and EBV) and Merkel cell polyomavirus (Merkel cell carcinoma of the skin). Co-factors may be involved. Immunization programmes focusing on hepatitis B and human papillomaviruses will reduce the incidence of liver and cervical cancer, respectively.
Pathology caused directly by microorganism
Direct effects may result from cell rupture, organ blockage or pressure effects
Organisms that multiply in cells and subsequently spread usually do so by rupturing the cell. Many viruses and some intracellular bacteria and protozoa behave in this way (Table 17.1). It is important to realize that many others do not. For example, viruses or bacteria may remain latent (e.g. herpes simplex virus and varicella-zoster virus in nerve ganglia, and Mycobacterium tuberculosis in macrophages), and many viruses can bud from a cell without disrupting it. The type of cell infected may also have an influence on survival of the organism. Thus although HIV causes lysis of CD4 T cells, macrophages are more resistant to lysis, perhaps because the virus activates the NFκB pathway, and so virus may persist within intracytoplasmic compartments. Other direct effects include:
• blockage of major hollow viscera by worms
• blockage of lung alveoli by dense growth of, e.g. Pneumocystis
• mechanical effects of large cysts (e.g. hydatid).
Table 17.1 Organisms that directly damage tissue
|
Organism |
Cell or tissue damaged |
Mechanism |
|
Viruses |
||
|
Poliovirus |
Neurones |
|
|
Bacteria |
||
|
Streptococcus mutans |
Teeth |
Acid production |
|
Mycobacteria |
Macrophages |
Damaged macrophage releases cytokines |
|
Fungi |
||
|
Histoplasma |
Macrophages |
Damaged macrophage releases cytokines |
|
Protozoa |
||
|
Plasmodium |
Erythrocytes |
Damaged erythrocyte removed |
|
Helminths |
||
|
Ascaris |
Intestinal occlusion |
Mechanical |
|
Biliary occlusion |
Mechanical, inflammation |
|
|
Echinococcus |
Hydatid cyst |
Pressure effects |

Many organisms directly damage or destroy the tissues they infect. This is especially common with cytopathic viruses. URT, upper respiratory tract.
Exotoxins are a common cause of serious tissue damage, especially in bacterial infection
The parasite may actively secrete ‘exotoxins’ (Table 17.2). In some cases, these are clearly part of its strategy for entry, spread or defence against the host, but sometimes they seem to be of little or no benefit to the parasite.
Table 17.2 Important exotoxins in disease
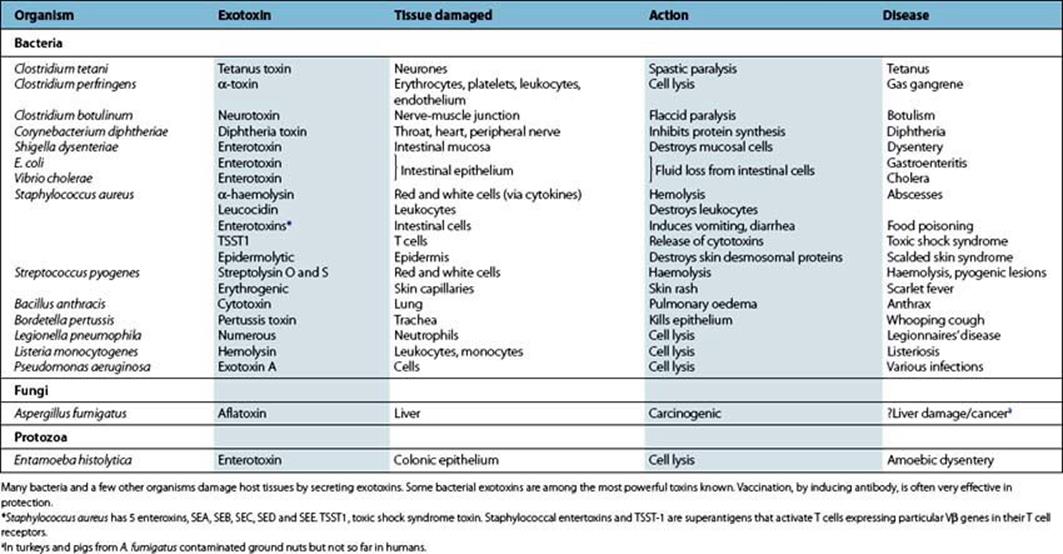
Most exotoxins are proteins and are often coded not by the bacterial DNA, but in plasmids (e.g. E. coli) or phages (e.g. botulism, diphtheria, scarlet fever). In some cases, they consist of two or more subunits, one of which is required for binding and entry to the cell while the other switches on or inhibits some cellular function.
Powerful toxins are generally secreted from extracellular microbes. Microbes that multiply in cells cannot afford to cause serious damage at too early a stage, and such toxins therefore tend to be less prominent in intracellular infections due to Mycobacteria, Chlamydia or Mycoplasma. For example, leprosy patients with lepromatous disease can live with huge bacterial loads for many years. Although many toxins can kill host cells, lower concentrations may be important by causing dysfunction in immune or phagocytic cells. For example, concentrations of streptolysin well below the cell-killing level will inhibit leukocyte chemotaxis, and the staphylococcal enterotoxin and epidermolytic toxins also have immunomodulatory activity at exceedingly low (nanogram to picogram) levels.
Inactivation of toxins without altering antigenicity results in successful vaccines
Toxins can often be inactivated (e.g. by formaldehyde) without altering their antigenicity, and the resulting toxoids are among the most successful of all vaccines (see Ch. 34), the classic examples being diphtheria and tetanus toxoids. Toxins are generally more highly conserved in their structure than the surface antigens of the organism secreting them. This allows for more effective cross-immunity and explains, for example, why scarlet fever (caused by streptococcal erythrotoxin) usually occurs only once, while streptococcal infections recur almost indefinitely.
Mode of action of toxins and consequences
These can be considered under five headings (Fig. 17.2).
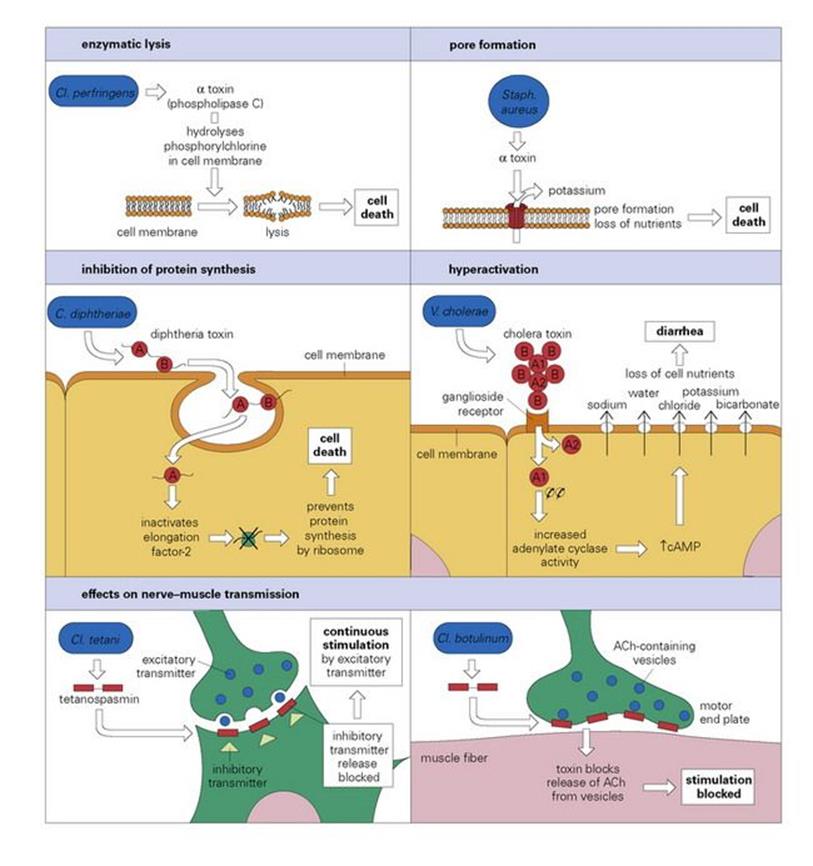
Figure 17.2 The mode of action of some exotoxins. Bacterial toxins act in a variety of ways. Often the toxin is a two-chain molecule, one chain being concerned with entry into cells while the other has inhibitory activity against some vital function. ACh, acetylcholine; cAMP, cyclic adenosine monophosphate; C, Corynebacterium; Cl, Clostridium; Staph, Staphylococcus; V, Vibrio.
Bacteria may produce enzymes to promote their survival or spread
A number of bacteria release enzymes that break down the tissues or the intercellular substances of the host, allowing the infection to spread freely. Among these enzymes are hyaluronidase, collagenase, DNase and streptokinase. Some staphylococci release a coagulase, which deposits a protective layer of fibrin onto and around the cells, thus localizing them.
Toxins may damage or destroy cells and are then known as haemolysins
Cell membranes can be damaged enzymatically by lecithinases or phospholipases, or by insertion of pore-forming molecules, which destroy the integrity of the cell. The collective term for such toxins is ‘haemolysins’, although many cells other than red blood cells can be affected. Both staphylococci and streptococci produce pore-forming toxins; pseudomonads release enzymatic haemolysins. The staphylococcal alpha-haemolysin is secreted as a soluble monomer but binds to a membrane protein to form a heptamer, making a beta-barrel pore in the membrane.
Toxins may enter cells and actively alter some of the metabolic machinery
Characteristically, these toxin molecules have two subunits. The A subunit is the active component, while the B subunit is a binding component needed to interact with receptors on the cell membrane. When binding occurs, the A subunit, or the whole toxin-receptor complex, is taken into the cell by endocytosis, and the A subunit becomes activated. Two well-studied toxins of this type are those of diphtheria (see Ch. 18) and cholera.
Diphtheria toxin blocks protein synthesis
Diphtheria toxin is synthesized as a single polypeptide and binds by the B subunit to target cells (Fig. 17.2). The polypeptide is partially cleaved and then the entire toxin-receptor complex is internalized. The A subunit then splits off and passes into the cytosol, where it inactivates the transfer of amino acids from transfer RNA to the polypeptide chain during translation of mRNA by ribosomes. It does this by catalysing attachment of adenosine diphosphate (ADP) ribose to the elongation protein (ADP ribosylation), effectively blocking protein synthesis.
Cholera toxin results in massive loss of water from intestinal epithelial cells
Cholera toxin is released as a complex of five B subunits surrounding the A subunit. The latter is cleaved into two fragments: A1 and A2, held by disulfide bonds. The B subunits bind to ganglioside receptors on intestinal epithelial cells, leading to internalization of the A subunits, which then separate from one another (Fig. 17.2). The Al portion then ADP-ribosylates one of the regulatory molecules involved in the production of cyclic adenosine monophosphate (cAMP). As a result, the regulatory molecule is unable to turn off cAMP production. The increased levels of cAMP in the cell change the sodium/chloride flux across the cell membrane, resulting in a massive outflow of water and electrolytes from the cell and causing the profuse diarrhea of cholera. The exotoxins of E. coli and salmonella have similar actions, as does pertussis toxin.
Tetanus and botulinum toxins are among the most potent affecting nerve impulses
These toxins are extremely potent and active at low doses. Tetanus and botulinum toxins have the characteristic A + B structure, the B subunit binding to ganglioside receptors on nerve cells. The internalized A subunit of tetanus is carried by axonal transport from the point of production to the central nervous system (CNS), where it interferes with synaptic transmission in inhibitory neurones by blocking neurotransmitter release. This allows the excitatory transmitter to continuously stimulate the motor neurones, causing spastic paralysis. Botulinum toxin enters the body via the intestine, escaping digestion and crossing the gut wall. The toxin affects peripheral nerve endings at the neuromuscular junction, blocking presynaptic release of acetylcholine. This prevents muscle contraction, causing flaccid paralysis.
Toxins as magic bullets
An interesting offshoot of the two-subunit structure of toxins is that by changing the specificity of the part responsible for attachment, the specificity of the toxin for a particular cell type can be changed. An example is the plant toxin ricin – the A subunit can be attached to a monoclonal antibody to make it a specific poison for tumour cells. The same strategy could obviously be used against parasites if desired.
Diarrhea
Diarrhea is an almost invariable result of intestinal infections
Diarrhea is one of the major causes of death in children worldwide, with rotavirus as the main culprit (see Ch. 22). In industrialized regions, bacterial pathogens such as Campylobacter and non-typhoidal Salmonella are increasingly important, and Clostridium difficile can be a problem in hospitals, particularly in the elderly. Diarrhea can be considered as:
• a means for the host to rid itself rapidly of the infectious organism
• a means for the infection to spread to other hosts.
Diarrhea is a feature of a wide range of organisms, but in only a few cases is the exact mechanism understood. While toxins are often the cause (e.g. cholera, shigella), microbial invasion and damage to epithelial cells may also be important. The pathophysiology, with changes in electron transport or loss of enterocytes, has been elucidated in some cases. Many of the organisms causing diarrhea can be ‘picked up’ from food, but the term ‘food poisoning’ is usually reserved for those cases where toxins are already present in the food rather than being generated during the growth of organisms in the intestine (Fig. 17.3). As would be expected, ‘food poisoning’ causes symptoms earlier – that is, hours after exposure rather than days (Table 17.3). Some viral infections, such as Norovirus, sometimes referred to as causing ‘winter vomiting disease’, cause outbreaks of diarrhea and vomiting, particularly in closed groups or communities – such as in hospitals or on cruise ships; in the UK in 2009/2010 there were 1888 reported hospital outbreaks, of which 1538 led to ward closures.
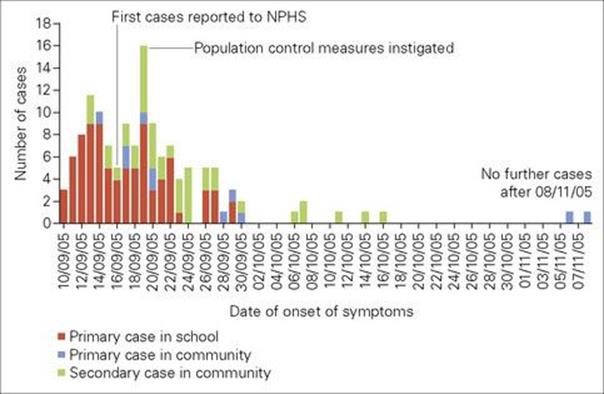
Figure 17.3 Outbreak of bloody diarrhea caused by Verocytotoxin-producing E. coli 0157 in South Wales, in 2005. The first cases had all eaten school dinners containing cooked meats from a single supplier. Of the total 157 reported cases, 65% were in school-aged children. Thirty-one people were admitted to hospital and one child died. NPHS National Public Health Service
(Redrawn from: The Public Inquiry into the September 2005 Outbreak of E. coli O157 in South Wales. Chairman H. Pennington, March 2009. http://wales.gov.uk/ecolidocs/3008707/reporten.pdf?skip=1&lang=en)
Table 17.3 Infectious causes of diarrhea
|
Onset |
Source |
|
|
Food poisoning (due to pre-formed toxin in food) |
||
|
Staphylococcus aureus |
1–6 h |
Cream, meat, poultry |
|
Clostridium perfringens |
8–20 h |
Reheated meat |
|
Clostridium botulinum |
12–36 h |
Canned food |
|
Bacillus cereus |
1–20 h |
Reheated foods |
|
Intestinal infections |
||
|
Rotavirus |
2–5 days |
Contact |
|
Norovirus |
1–2 days |
Contact (faecal–oral) |
|
Salmonella |
1–2 days |
Eggs |
|
Clostridium difficile |
1–2 days |
Faecal–oral |
|
Shigella |
1–4 days |
Faecal–oral |
|
Campylobacter |
1–4 days |
Poultry, domestic animals |
|
Vibrio cholerae |
2 days |
Faecal–oral |
|
Escherichia coli |
1–4 days |
Food |
|
Yersinia enterocolitica |
days–weeks |
Pets (e.g. dogs) |
|
Giardia lamblia |
1–2 weeks |
|
|
Cryptosporidium |
|
|
Worldwide, infectious diarrhea is the major cause of infant mortality.
Pathologic activation of natural immune mechanisms
Overactivity can damage host tissues
The very potent natural immune mechanisms discussed in Chapter 14 have inbuilt safety as far as specificity is concerned. They have had to evolve in the constant presence of the host’s ‘self’ antigens, to which they do not therefore respond. However, they are not so well controlled quantitatively, and there are many cases when overactivity not only damages an invading parasite, but also damages innocent host tissues. The expression of natural immunity often causes a certain amount of inflammation – and this can be severe, with tissue damage. Complement, polymorphs and tumour necrosis factor (TNF) play important roles.
Microbial endotoxin activates the immune system and induces cytokines, causing a bewildering variety of biologic effects (Fig. 17.4). At the clinical level, it can be responsible for septic shock.
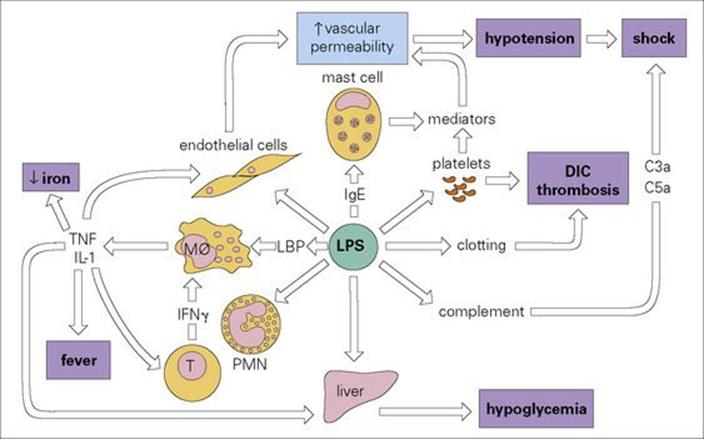
Figure 17.4 The many activities of bacterial endotoxin. Lipopolysaccharide (LPS) activates almost every immune mechanism as well as the clotting pathway and, as a result, LPS is one of the most powerful immune stimuli known. DIC, disseminated intravascular coagulation; IFN, interferon; IL, interleukin; LBP, LPS binding protein; Mφ, macrophage; PMN, polymorphonuclear leukocyte; TNF, tumour necrosis factor.
Endotoxins are typically lipopolysaccharides
‘Endotoxins’ of bacteria and other microorganisms have a deceptively similar name to exotoxins, but are profoundly different in their significance. Unlike exotoxins, these are integral parts of the microbial cell wall and are normally released only when the cell dies. Endotoxins are particularly characteristic of Gram-negative bacteria. A typical lipopolysaccharide (LPS) endotoxin is composed of:
• a conserved lipid portion (lipid A) inserted into the cell wall, responsible for much of the toxic activity
• a conserved core polysaccharide
• the highly variable O-polysaccharide, responsible for the serologic diversity which is a feature of organisms such as salmonellae and shigellae.
LPSs stimulate an extraordinary range of host responses – or perhaps one should say a wide range of responses have evolved to respond to LPSs. These include LPS binding protein (the LPS–LPS binding protein complex then binds to CD14 on macrophages and dendritic cells) and TLR4 (see Ch. 9). In the words of Lewis Thomas, ‘when we sense lipopolysaccharide, we are likely to turn on every defence at our disposal’ (Fig. 17.4). Evidently, the body needs to be aware of invading Gram-negative bacteria at the earliest possible stage.
Clinically, the most important effects of LPS are:
• fever
• vascular collapse (or shock).
As mentioned in Chapter 14, fever may benefit host or parasite, or both, and is currently considered to be mainly due to the action of two cytokines, interleukin 1 (IL-1) and tumour necrosis factor (TNF), on the hypothalamus. Both these cytokines are produced by macrophages in response to LPS (and to analogous molecules from other organisms, see below and Box 17.1).
![]()
Box 17.1  Lessons in Microbiology
Lessons in Microbiology
Is it a cold – or is it flu?
The common cold is usually caused by a rhinovirus, or a coronavirus. Real influenza, caused by the influenza virus usually has a more sudden onset and the combination of fever and a cough has a predictive value of around 80%. But what causes the symptoms of sore throat, sneezing, nasal discharge and nasal congestion?
Sore throat symptoms are thought to be caused by prostaglandins and bradykinin acting on sensory nerve endings in the airway. Sneezing is triggered by inflammatory mediators in the nose and nasopharynx acting on the trigeminal nerves. The plasma-rich exudate that forms part of the nasal discharge can change from clear to yellow/green during an upper respiratory infection. The colour reflects the recruitment of leukocytes into the airway lumen. If large numbers of leukocytes are present, the green protein myeloperoxidase found in the azurophil granules of neutrophils gives the discharge a green colour. Nasal congestion occurs later in infection, when inflammatory mediators such as bradykinin cause the large veins in the nasal epithelium to dilate. Common cold viruses do not cause such damage to the airway epithelium and infection may not create a cough – but influenza usually causes serious damage to the respiratory epithelium. Fever is mainly caused by the interleukins IL-1 and IL-6. It also seems that cytokines are responsible for muscle aches and pains, by causing the breakdown of muscle proteins. Of course, TNF was originally called cachexin, because of its ability to cause muscle wasting or cachexia.
Sometimes, in past flu epidemics, such as the Spanish flu epidemic in 1918, people died very quickly, within a few days of infection – which seems too fast for secondary infections to be responsible. Reconstructed viruses with the same haemagglutinin and neuraminidase seem to cause severe inflammation and it is possible that excessive cytokine release, in a ‘cytokine storm’, caused the pathology.
![]()
Endotoxin shock is usually associated with systemic spread of organisms
The commonest example of endotoxin (or ‘septic’) shock is septicaemia with Gram-negative bacteria such as E. coli or Neisseria meningitidis. However, many other organisms also release molecules that stimulate TNFα and/or IL-1 production (Table 17.4) and therefore function in part like LPS, although they are more or less unrelated in structure. In the ‘toxic shock syndrome’ of young women with staphylococcal infections of the genital tract, toxic shock syndrome toxin (TSST1) is the mediator; it acts as a superantigen, activating a large proportion of all T cells (up to 1 in 5, see Ch. 16) that express particular Vβ genes in their T cell receptors. Activating an enormous number of T cells produces enough cytokines to cause the toxic effect.
Table 17.4 Important endotoxins and functionally related molecules that induce TNF
|
Organisms |
Toxin |
|
Bacteria |
|
|
Gram-negative |
|
|
Gram-positive |
|
|
Staphylococcus aureus |
TSST1 |
|
Mycobacteria |
Lipoarabinomannan |
|
Bordetella pertussis |
Endotoxin |
|
Fungi |
|
|
Yeasts |
Zymosan |
|
Protozoa |
|
|
Plasmodium |
Phospholipids (exoantigens) |
Most endotoxins are lipopolysaccharides (LPS) and exert their main effects by stimulating cytokine release. TSST1, toxic shock syndrome toxin. LPS can also induce the secretion of other cytokines such as Interleukin-1.
Septic shock, however, is a complex phenomenon, and other bacterial components, such as peptidoglycans, may also play a part. Disseminated intravascular coagulation (DIC), hypoglycaemia and cardiovascular failure are all features of septic shock. In streptococcal infections, the culprits are pyrogenic (erythrogenic) exotoxins released by the bacteria.
The involvement of cytokines in the pathogenesis of shock is by no means a purely academic concern, because it suggests the possibility of treatment by antagonists of a small number of cytokines (e.g. by monoclonal antibodies or inhibitors), rather than by antibodies to the toxins themselves, which are of enormous antigenic diversity. This idea is discussed further in Chapter 35.
The cytokine most closely linked to disease is TNF
Raised concentrations of TNFα in the serum have been shown to correlate with severity in patients with meningococcal septicaemia and with Plasmodium falciparum malaria. However, animal experiments indicate that, in such cases, TNFα probably synergizes with other cytokines such as IL-1 and interferon-gamma (IFNγ), to produce its full effects. In meningococcal disease, TNFα concentrations in blood and cerebrospinal fluid (CSF) can change independently, the former being raised in septicaemia and the latter in meningitis; it therefore appears that the production and/or effects of TNFα can be restricted to a particular body compartment.
In some cases, it may be worth suppressing inflammation with steroids; e.g. a randomized trial in which dexamethasone was given to patients with acute bacterial meningitis showed that corticosteroid reduced mortality. The immune system itself also tries to control inflammation during sepsis by producing anti-inflammatory mediators such as IL-10 and TGFβ.
There may also be strain differences in the ability of bacteria to induce inflammation; Haemophilus influenzae strains isolated from patients with chronic obstructive pulmonary disease exacerbations induce more inflammation than colonizing strains not associated with the worsening of symptoms (Fig. 17.5).
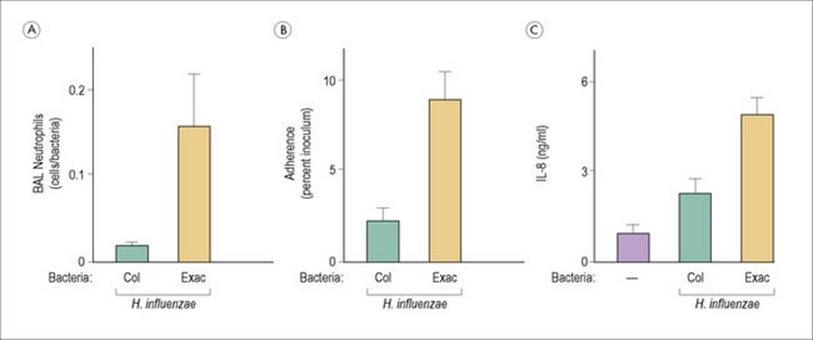
Figure 17.5 Haemophilus influenzae strains isolated from patients with chronic obstructive pulmonary disease (COPD) induce more inflammation than colonizing strains not associated with worsening of symptoms. H. influenzae strains from patients with COPD exacerbations (Exac) induce greater numbers of neutrophils (A), more adherence to airway epithelial cells (B) and more IL-8 (C) than isolates associated with colonization (Col). BAL, bronchoalveolar lavage.
(Redrawn from: Chin, C. L. et al. (2005) Haemophilus influenzae from patients with chronic obstructive pulmonary disease exacerbation induce more inflammation than colonizers. Am J Respir Crit Care Med 172:85–91.)
Complement is involved in several tissue-damaging reactions
The activation of complement is a vital part of immunity to many bacteria, viruses and protozoa (see Ch. 14). Complement can, however, be involved in tissue-damaging reactions, e.g. immune complex disease, which also involves antibody and, usually, polymorphonuclear leukocytes (PMNs). Complement also plays an important role in the acute inflammatory response by generating the chemotactic factors C3a and C5a (see Ch. 9). Animal experiments suggest that C5a contributes to cardiac problems during sepsis, as it binds to C5a receptors on cardiomyocytes (cardiomyocytes are also damaged by LPS itself and by inflammatory cytokines such as IL-1β, TNFα and IL-6).
Direct activation of complement by LPS may contribute to the shock induced by toxic amounts of this endotoxin, in which the levels of complement components (e.g. C3) drop profoundly; this response appears to involve both the classic and the alternative pathways, which are activated by the lipid and polysaccharide components, respectively. C3a and C5a are produced in large amounts, and there is frequently a severe decrease in the number of PMNs because of aggregation of these cells, adherence to vessel walls, and their activation to release toxic molecules, both oxidative and non-oxidative. When this occurs in the pulmonary capillaries, severe pulmonary oedema may result – the ‘adult respiratory distress syndrome’ (ARDS).
Disseminated intravascular coagulation is a rare but serious feature of bacterial septicaemia
Disseminated intravascular coagulation (DIC) can be a feature of bacterial (e.g. meningococcal) septicaemia, but is also seen in some virus infections such as Ebola fever (Ch. 28). The relative contributions of immune complexes, platelets, and direct activation of the clotting pathway via the effect of LPS on Hageman factor remain controversial. For example, the haemorrhagic phenomena of yellow fever are probably secondary to coagulation defects due to the extensive liver damage, whereas in dengue (‘haemorrhagic’) fever it has been suggested that there is immune complex deposition in blood vessels. However, in all these haemorrhagic syndromes the role of cytokines such as TNF also needs to be considered.
Mast cell degranulation in response to LPS is usually secondary to IgE antibody formation
Some insect venoms, however, may be able to activate mast cells directly, and reactions of this kind are called ‘anaphylactoid’.
Pathologic consequences of the immune response
Overreaction of the immune system is known as ‘hypersensitivity’
Adaptive immune responses are vital to defence against infection, as witnessed by the increased susceptibility to infectious disease of immunodeficient patients (see Ch. 30). The antimicrobial effects of lymphocyte responses act mainly by focusing or enhancing non-specific effector mechanisms (see Ch. 10). This may, however, also enhance the pathologic effects outlined above. The tissue-damaging effects of hypersensitivity are referred to as ‘immunopathologic’. Coombs and Gell classified hypersensitivity into four types in 1958, based on the immunologic mechanism underlying the tissue damaging reaction.
Each of the four main types of hypersensitivity can be of microbial or non-microbial origin
Hypersensitivity of microbial origin includes some of the most serious of these responses (Table 17.5). Organisms of many sorts can be involved, but one common feature is that the infection is prolonged, with continuous or repeated antigenic stimulation.
Table 17.5 Hypersensitivity of microbial origin
|
Coombs and Gell classification |
Principal mechanism |
Examples |
|
Type I (allergic/anaphylactic) |
IgE, mast cells |
Helminths |
|
Ascaris |
||
|
Hydatid (ruptured cyst) |
||
|
? Viral skin rash |
||
|
? Upper respiratory tract |
||
|
Viral infections |
||
|
Type II (cytotoxic) |
IgG to surface antigens |
Virus infected cells |
|
Malaria infected erythrocytes |
||
|
Autoantibodies in: |
||
|
Mycoplasma |
||
|
Streptococci |
||
|
Trypanosoma cruzi |
||
|
Type III (immune complex-mediated) |
Immune complexes |
In tissues: |
|
Allergic alveolitis |
||
|
Actinomycosis |
||
|
In blood vessels: |
||
|
Glomerulonephritis |
||
|
Malaria |
||
|
Streptococci |
||
|
Hepatitis B |
||
|
Syphilis |
||
|
Type IV (cell-mediated) |
T lymphocytes |
Granuloma |
|
Tuberculosis |
||
|
Leprosy (tuberculoid) |
||
|
Schistosomiasis (eggs) |
||
|
Histoplasma |
||
|
Mononuclear infiltration ± cell damage in many virus infections (i.e. tissue delayed-type hypersensitivity responses) with CD4 and CD8 T cell cytokines and macrophages playing roles |
||
|
Viral rashes |
||
|
Autoimmunity |
Cross-reaction with host |
Streptococcal myocarditis |
|
African trypanosomiasis |
All four classic types of hypersensitivity can be induced by infectious organisms, types II and III being the most commonly encountered. Note that some mechanisms mediating hypersensitivity also take part in protective immunity. PMN, polymorphonuclear leukocyte.
Type I hypersensitivity
These reactions are often called ‘immediate’, as they can occur within minutes, when the allergen triggers the degranulation of mast cells pre-coated with specific IgE antibodies.
Allergic reactions are a feature of worm infections
The most dramatic allergic (type I) reaction is that following the rupture of a hydatid cyst. Slow leakage of worm antigens ensures that the patient’s mast cells are sensitized with specific IgE, and the massive flood of antigens on rupture may cause acute fatal anaphylaxis, with vascular collapse and pulmonary oedema. Even the small amount of antigen used in diagnostic skin tests can have this effect, although this is rare.
Another worm associated with high levels of IgE is Ascaris, but here the pathologic consequences are mainly respiratory, with eosinophilic infiltrates and asthmatic episodes corresponding to passage of the parasite through the lung. The itching rashes characteristic of helminth infections when the worms die in the skin are probably also of this type, an example being ‘swimmer’s itch’ due to animal or avian schistosomes.
Why allergic reactions are such a feature of worm infections is not really clear, but they may be due to some feature of the antigens; in addition, it has been suggested that IgE plays a role in protection against worms. One would hope so, as in all other respects this class of antibody appears to be nothing but a nuisance.
Type II hypersensitivity
Type II reactions are mediated by antibodies to the infectious organism or autoantibodies
Strictly speaking, type II reactions are mediated by antibody (usually IgG) leading to cytotoxicity, either extracellular or intracellular (e.g. after phagocytosis). Antibody binds to the cell and, if complement is activated, the cell is lysed. Cytotoxicity by T cells is considered under type IV reactions. An important distinction can be made between antibodies to the (foreign) infectious organism and autoantibodies; the former kill host cells because they display foreign antigens, whereas the latter bind to unaltered host antigens, and both types of response occur in infectious disease (see Table 17.5). In the latter case, of course, the interesting question is why autoantibodies should be formed during infection, and several mechanisms have been postulated for this. However, the whole question of autoimmunity remains highly controversial.
In blood-stage malaria, microbial antigens attach themselves to host cells
It has been shown that the haemolytic anaemia of blood-stage malaria is due not to autoantibody, as previously thought, but to antibodies to parasite-derived antigens that have been picked up by red cells. In some cases, it may be the antigen–antibody complex that binds to the cell. A similar reaction can occur following quinine treatment of Plasmodium falciparum malaria – blackwater fever.
Antimyocardial antibody of group A β-haemolytic streptococcal infection is the classic autoantibody triggered by infection
This reaction is due to the presence of the same cross-reacting carbohydrate antigen on the bacterium and the myocardium. However, as more protein sequences are obtained and compared, numerous other similar examples have come to light, and it is possible that cross-reaction between microbial and human antigens may underlie a number of diseases of currently unknown origin. Whether this mimicry of host antigens has any survival value to the microbe is discussed in Chapter 16.
Type III hypersensitivity
Immune complexes cause disease when they become lodged in tissues or blood vessels
Immune complexes cause pathology if they are made in excess, if they are not removed properly from the circulation and if they deposit in tissues.
The formation of immune complexes can lead to phagocytosis and removal of antigen, but also to complement activation. Complications occur when the complexes escape removal by the phagocytes of the reticuloendothelial system and become lodged in the tissues or blood vessels, attracting complement and neutrophils. Release of lysosomal enzymes then results in local damage, which is particularly serious in small blood vessels, especially in the renal glomeruli. Immune complex disease is a major cause of both acute and chronic glomerulonephritis, and the majority of cases are probably the result of infection. There is also an important group in which autoantigen–autoantibody complexes are responsible (e.g. DNA–anti-DNA in systemic lupus erythematosus), but even these may ultimately be the consequence of a viral infection.
Like most other immunopathologic conditions, immune complex deposition is usually a feature of chronic infection (e.g. malaria). However, a persistent antigenic stimulus is not the only prerequisite, indicated by the fact that the most serious form of malarial nephropathy is found inPlasmodium malariae (quartan) malaria, which progresses despite successful treatment of the infection, while the nephropathy of P. falciparum (malignant tertian) malaria typically recovers after the infection has been cured. Predisposing factors may include a poor antibody response (in terms of amount or affinity), a particular tendency of the antigen itself to bind to vascular endothelium, or inhibition of the normal function of phagocytes or complement in removing circulating complexes.
Acute glomerulonephritis occurs as a serious complication of streptococcal infection (see Ch. 18) and is at least partly due to localization in glomeruli of immune complexes containing streptococcal antigens (see Fig. 17.7). Polymorph infiltration and alterations in the basement membrane cause leakage of albumin, even red cells, into the urine. The glomerulonephritis appears a few weeks after the infection has been terminated. When complexes are deposited over a long period (malarial nephropathy), the mesangial cell intrusions and fusion of foot processes cause a more irreversible impairment of glomerular function (chronic glomerulonephritis).
Occupational diseases associated with inhalation of fungi are the classic examples of immune complex deposition in the tissues
Immune complex deposition in the tissues, made famous by the work of Arthus on antigens injected into the skin of animals with pre-existing antibody (mainly IgG), manifests as a combination of thrombosis in small blood vessels and necrosis in the tissues due to PMN degranulation (Fig. 17.6). Perhaps the best-studied examples are the occupational diseases associated with inhalation of fungi (e.g. farmer’s lung, pigeon-fancier’s disease, maple bark stripper’s disease) in which chronic inflammation of the lung can lead to a state of destruction and fibrosis known as ‘extrinsic allergic alveolitis’, an unfortunate name since classic (IgE-mediated) allergy does not seem to be involved.
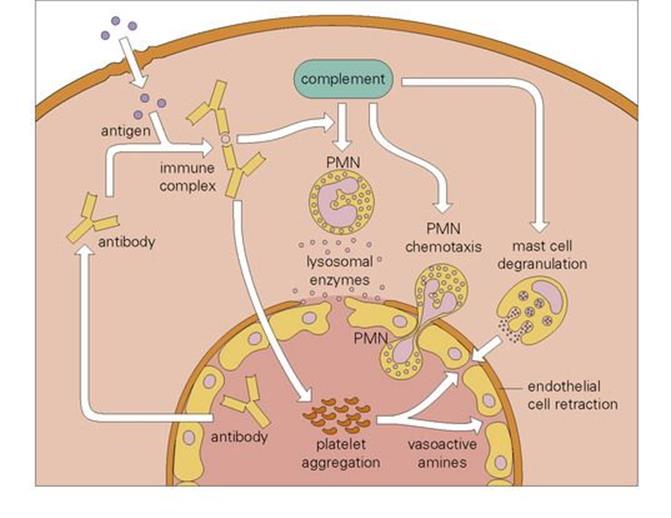
Figure 17.6 The Arthus reaction. Microbial antigens that enter the tissues (e.g. fungal particles in the lung) encounter antibody and form immune complexes. These activate complement and initiate chemotaxis of polymorphonuclear leukocytes (PMNs), and degranulation of these and tissue mast cells. The resulting inflammatory response is further potentiated by damage induced by PMN-derived lysosomal enzymes.
Another well-known model of immune complex disease is serum sickness
Serum sickness follows repeated injections of foreign protein, leading to circulating complexes, which deposit in the kidneys (Fig. 17.7), skin and joints. This was common in the pre-antibiotic days of passive serotherapy with horse serum for diphtheria (see Ch. 35). It is also a possible complication of treatment with murine monoclonal antibodies. As giving such blocking antibodies is an increasingly attractive approach to many conditions, the antibodies are now genetically engineered so that as much of the molecule as possible is humanized.
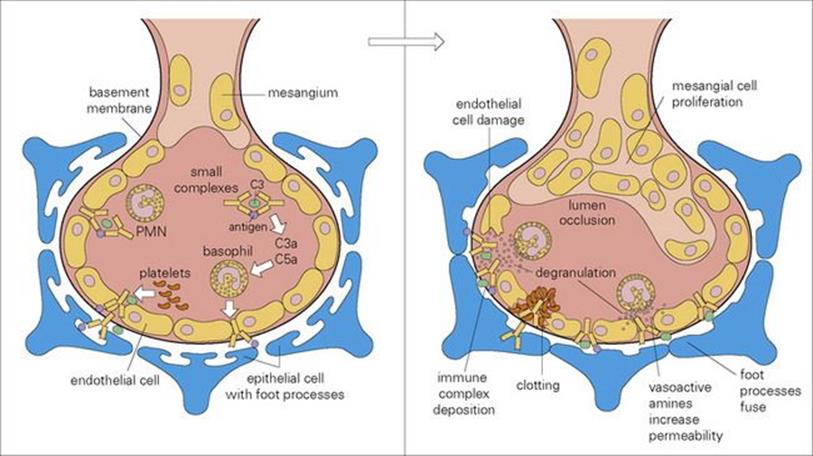
Figure 17.7 Glomerulonephritis caused by immune complex-mediated tissue damage. Type III hypersensitivity results in the deposition of immune complexes in the blood vessel walls, particularly at sites of high pressure, filtration or turbulence such as the kidney. Large complexes deposit on the glomerular basement membrane, while small ones pass through the basement membrane and then deposit on the epithelial side of the glomerulus. PMN, polymorphonuclear leukocyte.
Type IV hypersensitivity
Cell-mediated immune responses invariably cause some tissue destruction, which may be permanent
Despite the examples of antibody-mediated tissue damage discussed above, the antibody response generally achieves its purpose in eliminating invading organisms without any trace of damage to the host. Cell-mediated (type IV) responses are not quite so sure-footed, in that the activation of both T cells and macrophages invariably causes some tissue destruction, which may be reparable if not too prolonged, but can also lead to fibrosis and even calcification with serious permanent loss of tissue.
Confusion has occurred due to the use of a number of terms to describe and subdivide type IV responses. Some reflect actual pathologic conditions, while others describe the results of diagnostic skin tests, and none corresponds exactly to the processes by which cell-mediated immunity protects against infection (Table 17.6).
Table 17.6 Cell-mediated immunity in protection and disease
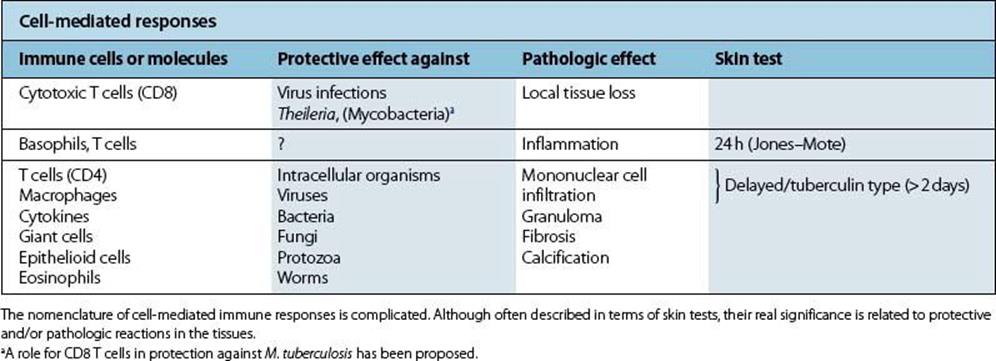
From the medical viewpoint, granuloma formation is the most important type IV response
The cell-mediated response to microbial antigen is responsible for granuloma formation and plays a major role in diseases such as tuberculosis, tuberculoid leprosy, lymphogranuloma inguinale, and in Toxocara infection. The complex involvement of the cytokine network in type IV responses poses certain paradoxes. For example, the tendency of some granulomas to undergo necrosis (e.g. caseation in tuberculosis) whereas others do not (e.g. leprosy, sarcoidosis) may be explained in terms of the different pattern of cytokines involved. TNF, often in association with some microbial products, is especially likely to cause necrosis through its effects on vascular endothelium, which probably accounts for much of its antitumour activity.
The clinical features of schistosomiasis are produced by cell-mediated immunity
The price paid for protective cell-mediated immunity is particularly well illustrated by the helminth disease schistosomiasis. Schistosoma mansoni (the blood fluke) lays eggs in the mesenteric venous system, some of which become lodged in small portal vessels in the liver. Strong cell-mediated reactions to secreted enzymes lead to granulomatous reactions around each egg, resulting in egg destruction and sparing of liver parenchyma from the toxic effects of the egg enzymes. However, the coalescent calcified granulomas ultimately cause portal cirrhosis, with portal hypertension, oesophageal varices and haematemesis (see Ch. 22).
The rather unexpected effect of malnutrition in reducing the incidence and severity of certain diseases (e.g. typhus, malaria) may be attributable to a reduction in immunopathology, though in the majority of diseases (e.g. measles, meningococcal infection, tuberculosis), the reverse is true. Indeed, poor nutrition is regarded as a major factor predisposing to the greater severity of many common infections in tropical countries.
Antibodies can also cause enhancement of pathology, as in dengue
Most cases of dengue haemorrhagic fever occur in people who get a second infection with the dengue virus. Antibody binding to the virus leads to greater internalization of virus, through Fc binding. The viral load falls as the fever falls, but this is when the most severe symptoms and pathology appear, including leakage of plasma from capillaries, haemorrhage and shock.
Skin rashes
A variety of skin rashes have an immunologic origin
The ways in which infections can affect the skin are detailed in Chapter 26, but here it should be mentioned that some rashes are considered to be immunologically mediated. For example, the characteristic skin rash of measles is absent in children with T-cell deficiency (e.g. thymic aplasia or DiGeorge syndrome), who instead develop a fatal systemic infection, indicating that the skin lesions are T-cell mediated and represent some form of successful cell-mediated immunity. In contrast, if children with T-cell deficiency are vaccinated with live vaccinia virus, they develop an inexorable spreading skin lesion, which is clearly a direct and not an immunopathologic effect.
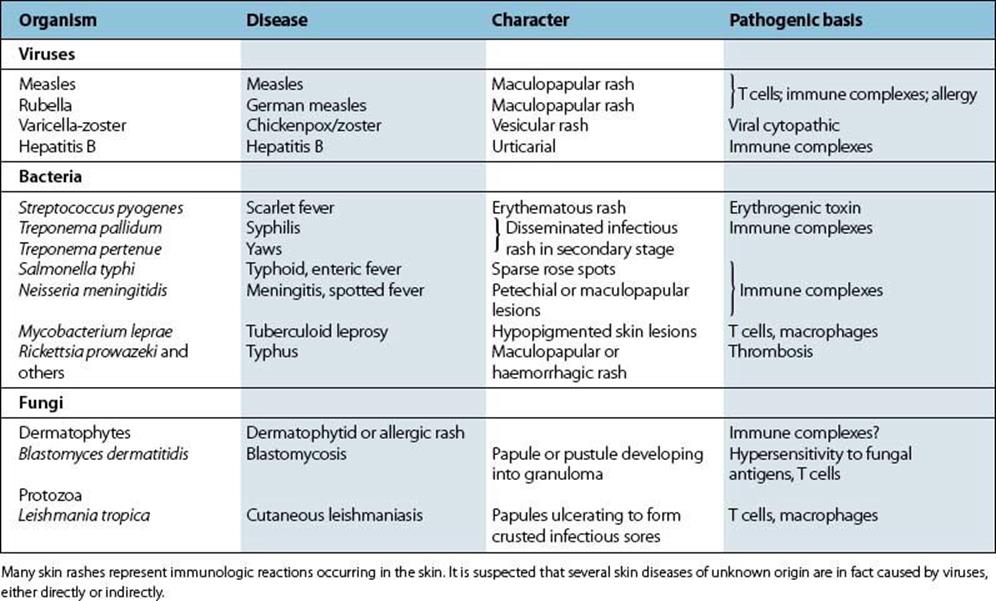
Table 17.7 lists the more common skin conditions of immunologic origin in which an infectious organism is thought to be involved. (Further details can be found in Ch. 26.)
Table 17.7 Skin rashes and their immunologic basis
The SARS coronavirus caused lung immunopathology and T-cell loss
The SARS coronavirus killed over 750 people in the 2002/3 epidemic. The lungs of patients with SARS viral pneumonia show diffuse damage to the alveoli, with the presence of multinucleate giant cells and many macrophages (Figure 17.8). The virus could also be found in other organs such as the intestine, liver and kidney of patients. The virus multiplies slowly at first, with a gradual increase in viral titre over the first 10 days. This suggests that the virus evades the innate immune response, possibly by failing to induce IFNα and IFNβ.
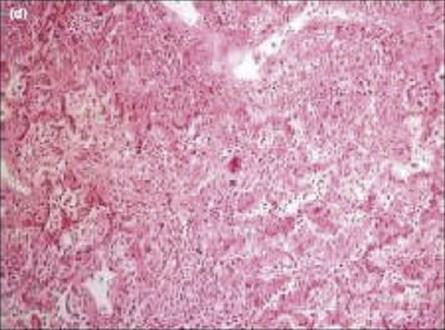
Figure 17.8 Pathology of SARS. The figure shows organizing diffuse alveolar damage, with giant cell formation in a patient who died of SARS.
(Courtesy of John Nicholls, Department of Pathology, The University of Hong Kong; taken from Lau, Y. L. and Peris, J. S. (2005) Pathogenesis of severe acute respiratory syndrome. Curr Opin Immunol 17:404, with permission).
During the acute phase of infection, lymphopenia occurs with loss of both CD4 and CD8 T cells. However, it was a puzzle as to how this could happen, as T cells do not express the angiotensin-converting enzyme 2, which is the functional receptor for the SARS virus. The answer seems to be that T-cell apoptosis is caused by proteins expressed by the virus.
The hygiene hypothesis – are we too clean?
Allergic diseases are more common than they used to be, and it has been proposed that this may be because most of us now grow up in an environment that is too clean. The hygiene hypothesis proposes that if we are exposed to a range of bacterial and viral infections in infancy, this may prevent the development of more harmful allergies by promoting a bias towards Th1 cytokine production. A more informative name might be ‘microbial exposure deficiency hypothesis’. Certainly, people living in Africa seem to have had more exposure to antigens, age for age, than those living in Europe, as they have more memory T cells and fewer naive T cells. Slightly surprisingly, it also seems that infections with helminths, which promote copious Th2 responses, also protect against development of atopy, possibly because they out-compete the allergen-specific IgE on the mast cells. Other factors, such as innate immunity and regulatory T cells that act to reduce harmful immune responses that cause immunopathology, may be involved.
Viruses and cancer
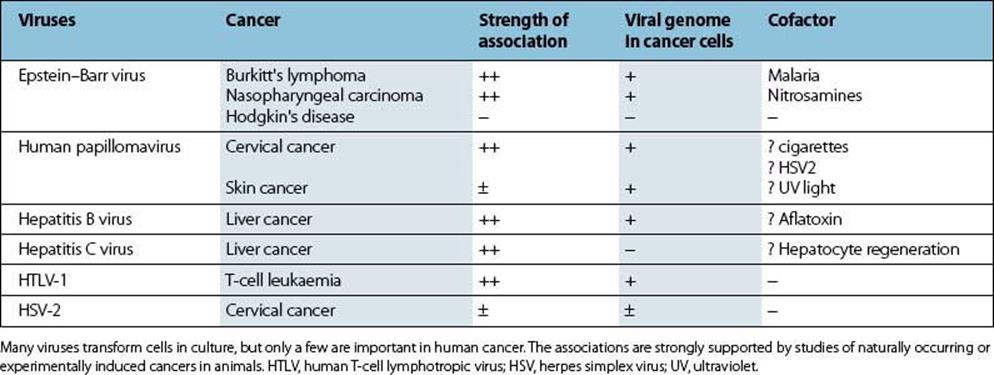
A variety of RNA and DNA viruses can cause permanent malignant changes within cells (Table 17.8). Such malignant transformation by these ‘tumour viruses’ has been extensively studied. An account of proviruses and oncogenes (genes causing malignancy) is included in Chapter 3. However, only a small number of human cancers have been shown to be associated with such tumour viruses (Table 17.9). Some of these include cancers associated with HIV and AIDS and are caused by the resulting immunosuppression and loss of immune control over members of the γ-herpesvirus family such as HHV-8 and EBV. There are latent and lytic components to their life cycles. Few genes are expressed in latent infections, allowing the virus to reside in specific sites with the potential for reactivation and setting up the environment for infected cells to undergo malignant change. The virus can disseminate in the lytic stage by release from infected cells. Some of the genes expressed can also promote tumour development. This part of the viral replicative cycle may be of more importance in virus-associated malignancy.
Table 17.8 Malignant transformation
|
Changes |
Details |
|
Morphology |
Loss of shape; rounding |
|
Decreased adhesion to surface |
|
|
Growth, contact |
Loss of contact inhibition of growth and movement |
|
Increased ability to grow from a single cell |
|
|
Increased ability to grow in suspension |
|
|
Capacity for continued growth (immortalization) |
|
|
Cellular properties |
DNA synthesis induced |
|
Chromosomal changes |
|
|
Appearance of new antigens (viral or cellular in origin) |
|
|
Biochemical properties |
Loss of fibronectin |
|
Reduced cAMP |
These changes occur when tumour viruses cause transformation of cultured cells. Many of these changes are obviously relevant for tumour production in vivo. cAMP, cyclic adenosine monophosphate.
Table 17.9 Viruses and human cancer
Human T-cell lymphotropic virus type 1 (HTLV-1) is associated with adult T-cell leukaemia/lymphoma
HTLV-1 and HTLV-2 are retroviruses that have no oncogenes (see Ch. 3). HTLV-1 proviral DNA is detectable in the cellular DNA of individuals with adult T-cell leukaemia/lymphoma (ATLL). Although reported around the world, HTLV-1 infection is endemic in Southern Japan, the Caribbean islands, West and Central Africa and parts of South America. Less is known about the geographic distribution of HTLV-2, which can be isolated from hairy T-cell leukaemia but has no association with malignancy. This virus can be found in certain Amerindian tribes and is associated with neurological and other chronic inflammatory conditions.
The carcinogenic nature of HTLV-1 is not due to activation of a cellular oncogene, but is due to the Tax accessory gene product enhancing transcription of host genes involved in cell division. This transactivation by Tax and stimulation of T-cell proliferation by HTLV-1 are thought to be central to oncogenesis. Adult T-cell leukemia/lymphoma (ATLL) cells contain the integrated HTLV-1 proviral DNA but the latter is not transcribed very actively. The TAX oncoprotein may cause precancerous cell proliferation, and HBZ, another oncoprotein, may maintain cell transformation. These infections are described in more detail in Chapter 26.
Epstein–Barr virus (EBV) is associated with nasopharyngeal carcinoma and lymphoma including post-transplant lymphoproliferative disease
Epstein–Barr virus is closely linked with the development of nasopharyngeal carcinoma (NPC) (see Ch. 18), which is common in Southern China and other parts of Asia (12–30 cases/100 000 people/year), less common in parts of North Africa, and rare elsewhere in the world. The reason for this restricted geographic distribution is unknown. There is no convincing evidence for specific carcinogenic EBV strains, but these effects could be due to the presence locally of co-carcinogens such as nitrosamines in salted fish. EBV DNA can be demonstrated in the cancer cells, but the precise mechanism for tumorigenicity is unknown; cellular oncogenes have not been implicated. People at high risk of developing NPC show high IgA titres to EBV capsid antigen a year or more before clinical symptoms appear.
Epstein–Barr virus is associated with Burkitt’s lymphoma
Burkitt’s lymphoma, a tumour of immature B cells, occurs in parts of East Africa, such as Uganda, and in Papua New Guinea in 6–14-year-old children, especially boys. EBV DNA is present in the tumour cells, but most of the many copies of the EBV gene are not integrated into the host cell DNA. The tumour is probably caused by the action of EBV on B cells, causing them to proliferate and making activation of cellular oncogenes more likely. The cellular oncogene c-myc is translocated from chromosome 8 to the immunoglobulin heavy chain locus on chromosome 14, where it is expressed. As a result of this, the B cell may be prevented from entering the resting stage. There is also down-regulation of adhesion and human leukocyte antigen (HLA) molecules, so that the EBV-containing cells, which are normally subject to immune control, develop into tumour cells. The Burkitt’s lymphoma cells also show other chromosomal abnormalities, but their role in tumorigenesis is unclear.
The fact that EBV is a common worldwide infection, whereas Burkitt’s lymphoma, like NPC, is strikingly localized geographically, points once again to the involvement of local co-factors, perhaps chemical or infectious co-carcinogens. Malaria is a recognized co-factor in Burkitt’s lymphoma, possibly having a role in altering the balance of the host’s immune response.
Epstein–Barr virus is also associated with Hodgkin’s lymphoma and lymphomas in immunosuppressed individuals
Epstein–Barr virus has been shown to be associated with classical Hodgkin’s lymphoma, in particular that seen in childhood and older adulthood. In addition, bearing in mind that cytotoxic T cells police EBV infection, the T-cell surveillance function is reduced in situations where the host is immunosuppressed, resulting in uncontrolled lymphoproliferation. This can be altered in EBV-driven post-transplant lymphoproliferative diseases by reducing the immunosuppression, although a consequence can be graft rejection. Other treatment is usually required, involving targeted monoclonal antibodies and cytotoxic chemotherapy.
EBV-associated primary cerebral lymphoma may occur in HIV-infected individuals. About 3% of patients with AIDS develop non-Hodgkin’s lymphomas, 20% of these occurring in the brain. The role of HIV is mostly indirect and is related to immunosuppression or B-cell activation. About 30% of AIDS-related lymphomas are Burkitt’s lymphomas.
Certain human papillomavirus infections are associated with cervical cancer
Papillomavirus infection is associated with a number of epithelial hyperproliferative diseases. The viral life cycle is intertwined with the host keratinocyte cells’ differentiation cycles. After small epithelial abrasions in skin or mucosal surfaces, these cells in the basal skin layer are exposed to HPV and the viral DNA becomes episomal and replicates with the host DNA using host synthetic machinery.
There are clear associations between the development of cervical cancer and infection with certain of the 77 distinct genotypes of human papillomavirus (HPV; see Chs 3, 21 and 26). They account for more than 80% of cervical cancers. Penile, vulval and rectal cancers are also associated with these types of HPV. Those at high risk include types 16 and 18, and those with low risk types 6 and 11. The latter cause cervical lesions but have a lower risk of progression to malignancy. HPV vaccine programmes were started in 2009 (see Ch. 34).
In most primary and metastatic cancer cells, the HPV genomes are present in integrated form (i.e. within the host genome), and certain viral oncoprotein genes referred to as E6 and E7 are transcribed and translated. Integration occurs at different chromosomal locations and the E6 and E7 open reading frames seem to be involved in transformation of epithelial cells and in maintenance of the transformed state, probably by binding to and inactivating tumour-suppressing cellular proteins concerned with regulation of the cell cycle. E6 is involved in turnover of p53, a tumour suppressor protein, and E7 binds and inactivates the retinoblastoma proteins. Both of these activities are critical in HPV-induced oncogenesis and result in genome instability, accumulation of oncogene mutations, uncontrolled cell growth and eventually cancer. Cervical cancer is an uncommon sequel to infection with these strains of HPV, and co-carcinogens such as cigarette smoke and herpes simplex virus (HSV) have been implicated.
Human papillomavirus infection is associated with squamous cell carcinoma of the skin
It is possible that ultraviolet light acts as a co-carcinogen, as is known to be the case with papillomaviruses and skin cancers in sheep and cattle. People with the rare autosomal recessive disease epidermodysplasia verruciformis are infected with up to 20 different but less common types of HPV, and 35% of patients develop multiple squamous cell carcinomas (SCC) of the skin. Of these tumours, 90% contain HPV-5 or HPV-8 DNA. These HPV types may act as co-carcinogens with ultraviolet light or immunosuppression in the development of non-melanoma skin cancers, the most common form of skin tumours in populations with fair skin.
HPVs may also play a role in the genesis of 90% of the skin cancers that appear in immunosuppressed organ transplant recipients, and cutaneous warts are common in these patients. In addition, there are reports that skin cancers in healthy individuals may be associated with HPV infection.
With respect to head and neck SCC, there are reports demonstrating the association of HPV-16 with SCC in the oral cavity and oropharynx and some evidence for HPV-18 resulting in oral cavity SCC.
Hepatitis B and hepatitis C viruses are major causes of hepatocellular carcinoma
The results, in sequential order, of chronic active hepatitis (CAH) include hepatocyte necrosis, chronic inflammation, cytokine production, fibrosis and finally cirrhosis. Therefore, CAH is a major driver for the development of hepatocellular carcinoma (HCC).
Individuals with active hepatitis B infections are 20-fold more likely to develop HCC than uninfected individuals. The oncogenic process depends on a number of predisposing factors that are both viral and host derived (Fig. 17.9). HCC is the outcome of chronic necroinflammatory liver disease associated with higher levels of HBV replication as well as the host immune response. Moreover, some HBV mutant strains and specific genotypes may be associated with HCC development. Integrated HBV sequences found in the HCC tumour cells may activate cellular oncogenes that encode proteins linked with controlling cell signalling, proliferation and viability, such as the myc family. Chronic inflammation, associated with increased liver cell proliferation, induces several rearrangements of the integrated HBV genome that can generate chromosomal instability. Moreover, there is evidence for the involvement of occult HBV infections, in which hepatitis B surface antigen cannot be detected but HBV DNA is integrated in the hepatocytes. Finally, HBV and HCV co-infection may act in concert with chronic alcohol consumption in liver carcinogenesis.
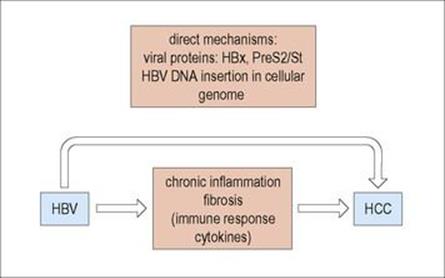
Figure 17.9 Development of hepatocellular carcinoma (HCC).
HCC is more common in certain parts of the world, such as Africa and South-East Asia, and this may be due to the presence of co-carcinogens (e.g. aflatoxin). However, the closely related hepadnavirus of woodchucks (Box 17.2) causes the same tumour in these animals in the apparent absence of co-carcinogens.
![]()
Box 17.2 Lessons in Microbiology
The many faces of hepatitis B
Classic epidemiologic studies on hepatitis B virus in Taiwan showed two things. First, 90% of those infected in infancy became carriers, as did 23% of those infected at 1–3 years, but only 3% of those infected as university students. Second, among 3454 carriers of HBsAg, there were 184 cases of hepatocellular carcinoma, whereas there were only 10 cases among 19 253 non-carriers. Some 80% of all liver cancers are due to hepatitis B.
Worldwide, there are about 350 million carriers of this virus and, therefore, with liver cancer causing up to 2 million deaths each year, hepatitis B virus is second only to tobacco as a human carcinogen.
The mechanism of carcinogenesis is not clear. Nearly all human cancers show chromosomal integration of the virus, but there is great variation in integration site and in the number of copies of the viral genome.
Very similar viruses infect woodchucks, ground squirrels and Pekin ducks. In Northwest USA, 30% of woodchucks are carriers and most develop liver cancer in later life. In this host, the virus infects not only liver cells but also lymphoid cells in the spleen, peripheral blood and thymus, and pancreatic acinar cells and bile duct epithelium.
![]()
The mechanism by which hepatitis C virus (HCV) causes HCC is considered to be indirect, as HCV sequences are not integrated into tumour cells. It is thought that the persistent hepatocyte damage and inflammation in HCV carriers, together with the effects of cytokines on the development of fibrosis and hepatocyte proliferation, results in HCC. It is also thought that HCV may have a direct action via specific viral proteins interacting with host cell factors modulating pathways such as cell signalling and proliferation and apoptosis that result in malignant transformation of liver cells. Once cirrhosis is established, there is a 1–4% risk of HCC. In addition, HCV is associated with mixed cryoglobulinaemia, a lymphoproliferative disorder that can develop into B-cell non-Hodgkin’s lymphoma.
Several DNA viruses can transform cells in which they are unable to replicate
In addition, the viral genome is sometimes integrated into the host cell genome. Extensive studies have been carried out with the conclusion that despite high oncogenicity in vitro and in laboratory animals, these viruses do not seem to be important in human cancer. For instance:
• Human adenoviruses transform cells in culture and cause sarcomas experimentally in hamsters. About 10% of the adenovirus genome integrates, and the T antigen is expressed. However, adenoviruses are not associated with human cancer.
• Polyomavirus (Latin: poly, many; oma, tumours), a mouse papovavirus, and simian vacuolating virus 40 (SV40), a monkey papovavirus, both cause tumours in experimentally inoculated hamsters. The viral DNA is integrated into tumour cells, and T antigens are expressed. Are these viruses, or their human equivalents (BK and JC viruses), linked with human cancers? An incident occurred about 30 years ago, when thousands of children were accidentally inoculated with SV40 virus present in certain batches of poliovirus vaccine. The formalin inactivation procedure had failed to kill the SV40 virus present in the monkey kidney cells in which the polio vaccine had been grown. There was, however, no consequent increase in tumour incidence in the SV40-infected individuals. Nevertheless, evidence is accumulating that JC, BK and SV40 viruses associated with certain cancers of the brain, with certain lymphomas and with other tumours. However, a causative role has not been established.
Kaposi’s sarcoma is caused by HHV-8
Kaposi’s sarcoma (KS) is a multicentric tumour that involves massive proliferation of endothelial cells. It is 300 times more common among patients with AIDS than among other immunosuppressed groups, but is seen almost entirely in those who acquired HIV by sexual contact. It was identified in 1994 from a lesion in an individual with AIDS-associated KS. Human herpesvirus 8 (HHV-8), referred to originally as the Kaposi’s sarcoma-associated herpesvirus (KSHV), appears to be sexually transmitted and is present in the tumours.
HHV-8 latently infects most tumour cells in lymphomas and KS. As a result, they are resistant to antiviral drugs targeting herpesviruses in the lytic cycle of replication. Novel treatment strategies are being investigated that induce HHV-8 to enter the lytic part of the life cycle leading to apoptosis of HHV-8 infected cells. It has been demonstrated that one of the HHV-8 lytic genes encodes the viral G-protein-coupled receptor (vGPCR), a constitutively active cellular chemokine receptor. vGPCR signalling can result in cell proliferation, the production of angiogenic factors and, in an animal model, can lead to KS-like lesions (Fig. 17.10). Moreover, there are indirect mechanisms involved in oncogenesis relating to altered T-cell responses and HHV-8 immunoregulation.
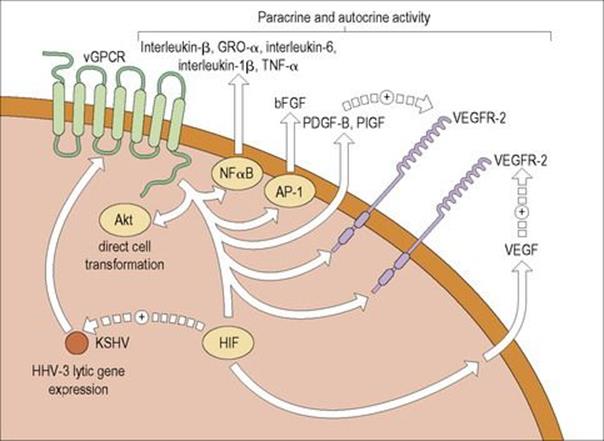
Figure 17.10 Activities of the vGPCR protein in HHV-8. The constitutively active viral G-protein-coupled receptor (vGPCR) of HHV-8 may promote the development of Kaposi’s sarcoma by means of a variety of mechanisms. Signalling by vGPCR activates Akt, an activated protein kinase which directly induces cell transformation. The vGPCR also results in the production of a variety of other factors, including the nuclear factor (NF)-κB-dependent factors interleukin-8, growth-related protein α (GRO-α), interleukin-6, interleukin-1β and tumour necrosis factor alpha (TNFα); AP-1-dependent basic fibroblast growth factor (bFGF); platelet-derived growth factor B (PDGF-B); and placental growth factor (PIGF). Some, but not all, studies have found that vGPCR induces secretion of vascular endothelial growth factor (VEGF) and there is evidence that this secretion may be mediated by hypoxia-inducible factor (HIF). Also, vGPCR up-regulates the expression of the VEGF receptors 1 and 2 (VEGFR-1 and VEGFR-2, respectively). PIGF, VEGF and other factors can act in an autocrine or paracrine fashion to promote Kaposi’s sarcoma.
(Redrawn from: Yarchoan, R. (2006) Key role for a viral lytic gene in Kaposi’s sarcoma. N Engl J Med 355:1383–1385, with permission.)
The incidence of KS has fallen sharply in HIV-infected individuals after the advent of highly active antiretroviral therapy (HAART), an element of which is thought to be due to direct activity on KS by these agents.
HHV-8 is also associated with benign and malignant lymphomatous conditions, namely multicentric Castleman’s disease and primary effusion lymphoma, respectively.
Bacteria associated with cancer
The association between Helicobacter pylori and stomach and duodenal cancer, including gastric mucosa-associated lymphoid tissue (MALT) lymphoma, is referred to in Chapter 22. It is thought that a number of inflammatory reactions are triggered as a result of H. pylori colonizing the stomach mucosa leading to chronic atrophic gastritis (CAG). This sets off a cascade of mucosal changes resulting in intestinal metaplasia, dysplasia and carcinoma. The question is whether there are other environmental or genetic co-factors involved in oncogenesis. The tumour is associated with chronic inflammation secondary to H. pylori colonization, but it is thought that the bacterium alone is not sufficient for cancer to develop.
![]()
 Key Facts
Key Facts
• Tissue damage or disease can be caused by infectious organisms in several ways.
• Infectious organisms may destroy cells directly (e.g. cytopathic viruses), release toxins that destroy cells or their cellular function (e.g. staphylococcus or tetanus toxins), overstimulate normal defence systems (e.g. LPS) or stimulate excessive or prolonged adaptive responses.
• Such effects of infectious organisms on defence systems may be antibody- or T-cell-mediated and are collectively known as ‘hypersensitivity reactions’ or ‘immunopathology’.
• Some viruses have been shown to be involved in the initiation of tumours, with the viral genome being found in the cancer cells. The restricted geographic distribution of some of these tumours may be due to the local presence of co-carcinogens.
![]()