Harper’s Illustrated Biochemistry, 29th Edition (2012)
SECTION VI. Special Topics
Chapter 50. Plasma Proteins & Immunoglobulins
Robert K. Murray, MD, PhD, Molly Jacob, MB BS, MD, PhD, & Joe Varghese, MB BS, MD
OBJECTIVES
After studying this chapter, you should be able to:
![]() List the major functions of blood.
List the major functions of blood.
![]() Explain the functions of the major plasma proteins, including albumin, haptoglobin, transferrin, ceruloplasmin, α1-antitrypsin, and α2-macroglobulin.
Explain the functions of the major plasma proteins, including albumin, haptoglobin, transferrin, ceruloplasmin, α1-antitrypsin, and α2-macroglobulin.
![]() Describe how iron homeostasis is maintained and how it is affected in certain disorders.
Describe how iron homeostasis is maintained and how it is affected in certain disorders.
![]() Describe the general structures and functions of the five classes of immunoglobulins and the uses of monoclonal antibodies.
Describe the general structures and functions of the five classes of immunoglobulins and the uses of monoclonal antibodies.
![]() Appreciate that the complement system is involved in a number of important biological processes.
Appreciate that the complement system is involved in a number of important biological processes.
![]() Indicate the causes of Wilson disease, Menkes disease, the lung and liver diseases associated with α1-antitrypsin deficiency, amyloidosis, multiple myeloma, and agammaglobulinemia.
Indicate the causes of Wilson disease, Menkes disease, the lung and liver diseases associated with α1-antitrypsin deficiency, amyloidosis, multiple myeloma, and agammaglobulinemia.
BIOMEDICAL IMPORTANCE
The fundamental role of blood in the maintenance of homeostasis (see Chapter 51) and the ease with which blood can be obtained have meant that the study of its constituents has been of central importance in the development of biochemistry and clinical biochemistry. The basic properties of a number of plasma proteins, including the immunoglobulins (antibodies), are described in this chapter. Changes in the amounts of various plasma proteins and immunoglobulins occur in many diseases and can be monitored by electrophoresis or other suitable procedures. As indicated in an earlier chapter, alterations of the activities of certain enzymes found in plasma are of diagnostic use in a number of pathologic conditions. Plasma proteins involved in blood coagulation are discussed in Chapter 51.
THE BLOOD HAS MANY FUNCTIONS
The functions of blood—except for specific cellular ones such as oxygen transport and cell-mediated immunologic defense—are carried out by plasma and its constituents (Table 50-1).
TABLE 50–1 Major Functions of Blood
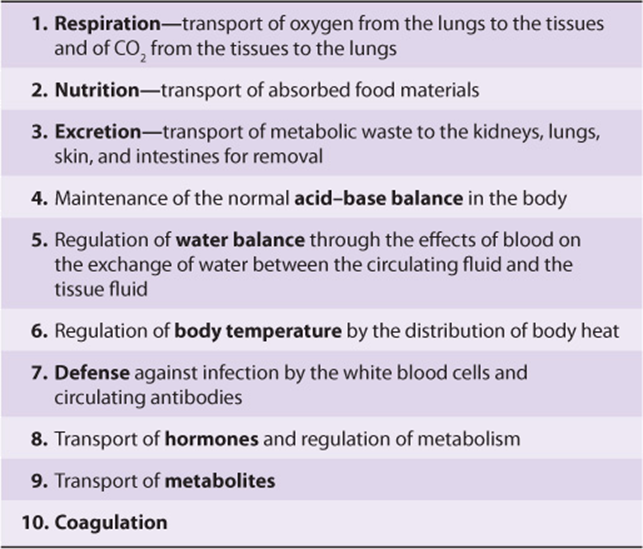
Plasma consists of water, electrolytes, metabolites, nutrients, proteins, and hormones. The water and electrolyte composition of plasma is practically the same as that of all extracellular fluids. Laboratory determinations of levels of Na+, K+, Ca2+, Mg2+, Cl–, ![]() , PaCO2, and of blood pH are important in the management of many patients.
, PaCO2, and of blood pH are important in the management of many patients.
PLASMA CONTAINS A COMPLEX MIXTURE OF PROTEINS
The concentration of total protein in human plasma is approximately 7.0-7.5 g/dL and comprises the major part of the solids of the plasma. The proteins of the plasma are actually a complex mixture that includes not only simple proteins but also conjugated proteins such as glycoproteins and various types of lipoproteins. Use of proteomic techniques is allowing the isolation and characterization of previously unknown plasma proteins, some present in very small amounts (eg, detected in hemodialysis fluid and in the plasma of patients with cancer), thus expanding the plasma proteome. Thousands of antibodies are present in human plasma, although the amount of any one antibody is usually quite low under normal circumstances. The relative dimensions and molecular masses of some of the most important plasma proteins are shown in Figure 50–1.

FIGURE 50–1 Relative dimensions and approximate molecular masses of protein molecules in the blood (Oncley).
The separation of individual proteins from a complex mixture is frequently accomplished by the use of solvents or electrolytes (or both) to remove different protein fractions in accordance with their solubility characteristics. This is the basis of the so-called salting-out methods, which find some usage in the determination of protein fractions in the clinical laboratory. Thus, one can separate the proteins of the plasma into three major groups—fibrinogen, albumin, and globulins—by the use of varying concentrations of sodium or ammonium sulfate.
The most common method of analyzing plasma proteins is by electrophoresis. There are many types of electrophoresis, each using a different supporting medium. In clinical laboratories, cellulose acetate is widely used as a supporting medium. Its use permits resolution, after staining, of plasma proteins into five bands, designated albumin, α1, α2, β, and γ fractions, respectively (Figure 50–2). The stained strip of cellulose acetate (or other supporting medium) is called an electrophoretogram. The amounts of these five bands can be conveniently quantified by use of densitometric scanning machines. Characteristic changes in the amounts of one or more of these five bands are found in many diseases.
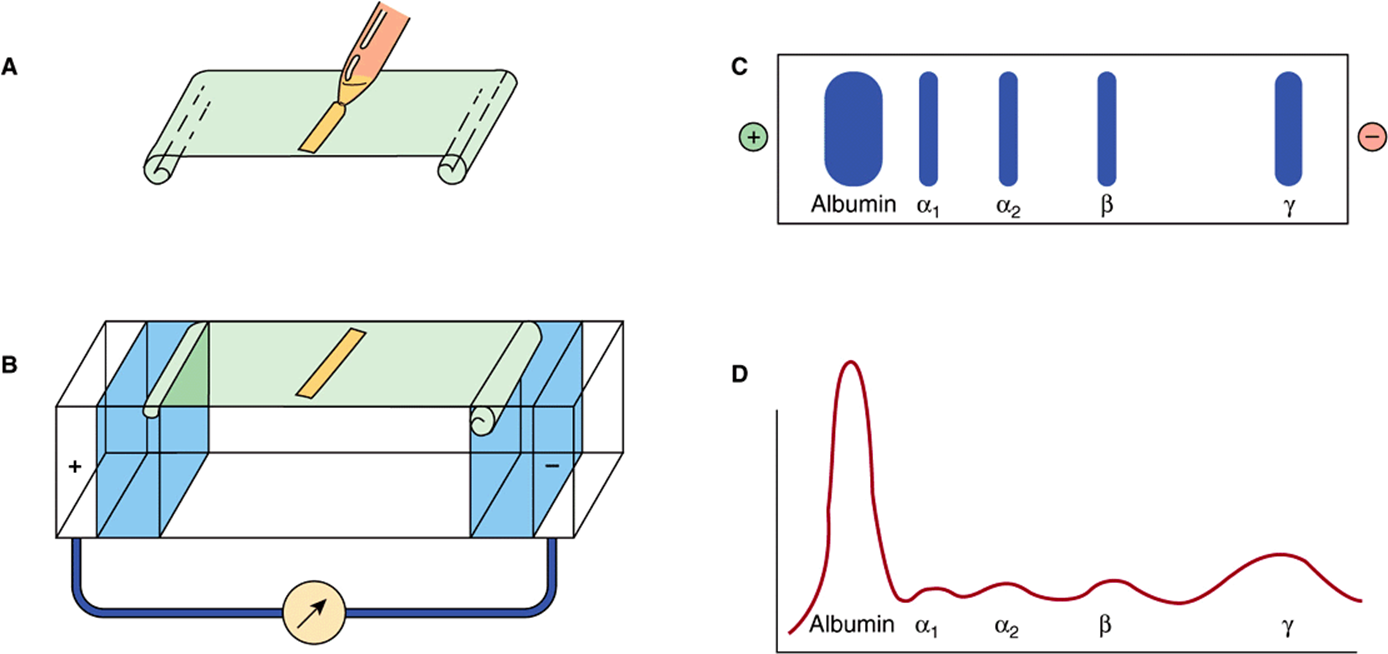
FIGURE 50–2 Technique of cellulose acetate zone electrophoresis. (A) A small amount of serum or other fluid is applied to a cellulose acetate strip. (B) Electrophoresis of sample in electrolyte buffer is performed. (C) Separated protein bands are visualized in characteristic positions after being stained. (D) Densitometer scanning from cellulose acetate strip converts bands to characteristic peaks of albumin, α1,-globulin, β2-globulin, β-globulin, and γ-globulin. (Reproduced, with permission, from Parslow TG et al (editors): Medical Immunology, 10th ed. McGraw-Hill, 2001.)
The Concentration of Protein in Plasma Is Important in Determining the Distribution of Fluid Between Blood & Tissues
In arterioles, the hydrostatic pressure is about 37 mmHg, with an interstitial (tissue) pressure of 1 mmHg opposing it. The osmotic pressure (oncotic pressure) exerted by the plasma proteins is approximately 25 mmHg. Thus, a net outward force of about 11 mmHg drives fluid out into the interstitial spaces. In venules, the hydrostatic pressure is about 17 mmHg, with the oncotic and interstitial pressures as described above; thus, a net force of about 9 mmHg attracts water back into the circulation. The above pressures are often referred to as the Starling forces. If the concentration of plasma proteins is markedly diminished (eg, due to severe protein malnutrition), fluid is not attracted back into the intravascular compartment and accumulates in the extravascular tissue spaces, a condition known as edema. Edema has many causes; protein deficiency is one of them.
Plasma Proteins Have Been Studied Extensively
Because of the relative ease with which they can be obtained, plasma proteins have been studied extensively in both humans and animals. Considerable information is available about the biosynthesis, turnover, structure, and functions of the major plasma proteins. Alterations of their amounts and of their metabolism in many disease states have also been investigated. In recent years, many of the genes for plasma proteins have been cloned and their structures determined.
The preparation of antibodies specific for the individual plasma proteins has greatly facilitated their study, allowing the precipitation and isolation of pure proteins from the complex mixture present in tissues or plasma. In addition, the use of isotopes has made possible the determination of their pathways of biosynthesis and of their turnover rates in plasma.
The following generalizations have emerged from studies of plasma proteins.
Most Plasma Proteins Are Synthesized in the Liver
This has been established by experiments at the whole animal level (eg, hepatectomy) and by use of the isolated perfused liver preparation, of liver slices, of liver homogenates, and of in vitro translation systems using preparations of mRNA extracted from liver. However, the γ-globulins are synthesized in plasma cells and certain plasma proteins are synthesized in other sites, such as endothelial cells.
Plasma Proteins Are Generally Synthesized on Membrane-Bound Polyribosomes
They then traverse the major secretory route in the cell (rough endoplasmic membrane → smooth endoplasmic membrane → Golgi apparatus → secretory vesicles) prior to entering the plasma. Thus, most plasma proteins are synthesized as preproteins and initially contain amino terminal signal peptides (Chapter 46). They are usually subjected to various posttranslational modifications (proteolysis, glycosylation, phosphorylation, etc) as they travel through the cell. Transit times through the hepatocyte from the site of synthesis to the plasma vary from 30 min to several hours or more for individual proteins.
Most Plasma Proteins Are Glycoproteins
Accordingly, they generally contain either N- or O-linked oligosaccharide chains, or both (Chapter 47). Albumin is the major exception; it does not contain sugar residues. The oligosaccharide chains have various functions (Table 47-2). Removal of terminal sialic acid residues from certain plasma proteins (eg, ceruloplasmin) by exposure to neuraminidase can markedly shorten their half-lives in plasma (Chapter 47).
Many Plasma Proteins Exhibit Polymorphism
A polymorphism is a Mendelian or monogenic trait that exists in the population in at least two phenotypes, neither of which is rare (ie, neither of which occurs with frequency of <1-2%). The ABO blood group substances (Chapter 52) are the best known examples of human polymorphisms. Human plasma proteins that exhibit polymorphism include α1- antitrypsin, haptoglobin, transferrin, ceruloplasmin, and immunoglobulins. The polymorphic forms of these proteins can be distinguished by different procedures (eg, various types of electrophoresis or isoelectric focusing), in which each form may show a characteristic migration. Analyses of these human polymorphisms have proved to be of genetic, anthropologic, and clinical interest.
Each Plasma Protein Has a Characteristic Half-Life in the Circulation
The half-life of a plasma protein can be determined by labeling the isolated pure protein with 131I or Cr51 under mild, nonde-naturing conditions. The labeled protein is freed of unbound free isotope and its specific activity (disintegrations per minute per milligram of protein) determined. A known amount of the radioactive protein is then injected into a normal adult subject, and samples of blood are taken at various time intervals for determinations of radioactivity. The values for radioactivity are plotted against time, and the half-life of the protein (the time for the radioactivity to decline from its peak value to one-half of its peak value) can be calculated from the resulting graph, discounting the times for the injected protein to equilibrate (mix) in the blood and in the extravascular spaces. The half-lives obtained for albumin and haptoglobin in normal healthy adults are approximately 20 and 5 days, respectively. In certain diseases, the half-life of a protein may be markedly altered. For instance, in some gastrointestinal diseases such as regional ileitis (Crohn’s disease), considerable amounts of plasma proteins, including albumin, may be lost into the bowel through the inflamed intestinal mucosa. Patients with this condition have a protein-losing gastroenteropathy, and the half-life of injected iodinated albumin in these subjects may be reduced to as little as 1 day.
The Levels of Certain Proteins in Plasma Increase During Acute Inflammatory States or Secondary to Certain Types of Tissue Damage
These proteins are called “acute-phase proteins” (or reactants) and include C-reactive protein (CRP, so named because it reacts with the C polysaccharide of pneumococci), α1-antitrypsin, haptoglobin, α1 acid glycoprotein, and fibrinogen. The elevations of the levels of these proteins vary from as little as 50% to as much as 1000-fold in the case of CRP. Their levels are also usually elevated during chronic inflammatory states and in patients with cancer. These proteins are believed to play a role in the body’s response to inflammation. For example, C-reactive protein can stimulate the classic complement pathway (see below), and α1-antitrypsin can neutralize certain proteases released during the acute inflammatory state. CRP is used as a marker of tissue injury, infection, and inflammation, and there is considerable interest in its use as a predictor of certain types of cardiovascular conditions secondary to atherosclerosis. The cytokine (= a protein made by cells that affects the behavior of other cells) interleukin-1 (IL-1), a poly-peptide released from mononuclear phagocytic cells, is the principal—but not the sole—stimulator of the synthesis of the majority of acute phase reactants by hepatocytes. Additional molecules such as IL-6 are involved, and they as well as IL-1 appear to work at the level of gene transcription.
Nuclear factor kappa-B (NFkB) is a transcription factor that has been involved in the stimulation of synthesis of certain of the acute phase proteins. This important factor is also involved in the expression of many cytokines, chemokines, growth factors, and cell adhesion molecules implicated in immunologic phenomena. Normally, it exists in an inactive form in the cytosol but is activated and translocated to the nucleus via the action of a number of molecules (eg, interleukin-1) produced in processes such as inflammation, infection, and radiation injury.
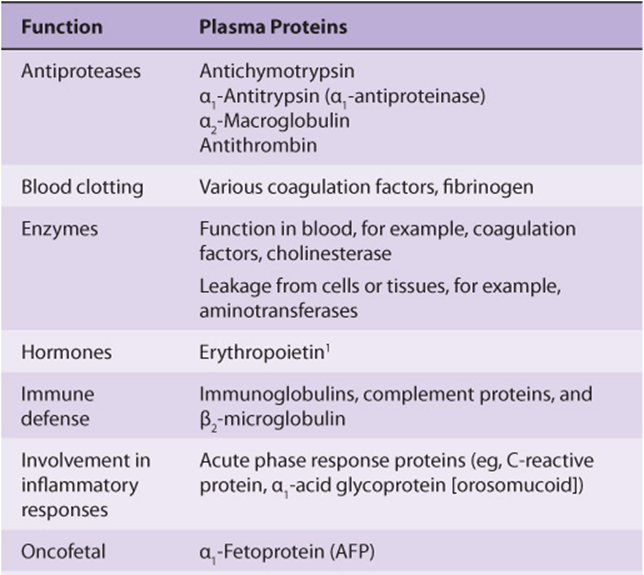
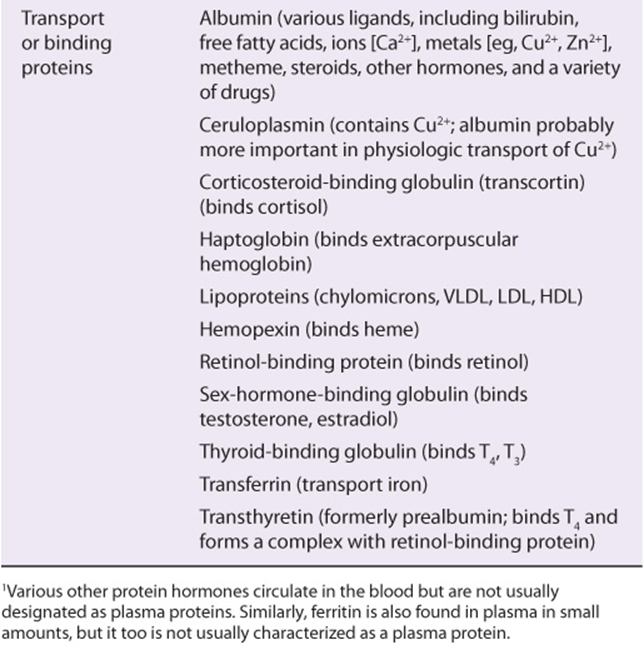
Table 50-2 summarizes the functions of many of the plasma proteins. The remainder of the material in this chapter presents basic information regarding selected plasma proteins: albumin, haptoglobin, transferrin, ceruloplasmin, α1-antitrypsin, α2-macroglobulin, the immunoglobulins, and the complement system. The lipoproteins are discussed in Chapter 25. New information is constantly forthcoming on plasma proteins and their variants (including those discussed here), as the techniques of proteomics, particularly sensitive new methods of determining proteins sequences by mass spectrometry (see Chapter 4), are applied to their study. A number of laboratories are participating in efforts to determine the complete human plasma protein proteome. It is believed that this will shed further light on genetic variations in humans and also provide many new biomarkers to aid in the diagnosis of many diseases. (A biomarker has been defined as a characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention.)
TABLE 50–2 Some Functions of Plasma Proteins
Albumin Is the Major Protein In Human Plasma
Albumin (69 kDa) is the major protein of human plasma (3.44.7 g/dL) and makes up approximately 60% of the total plasma protein. About 40% of albumin is present in the plasma, and the other 60% is present in the extracellular space. The liver produces about 12 g of albumin per day, representing about 25% of total hepatic protein synthesis and half its secreted protein. Albumin is initially synthesized as a preproprotein. Its signal peptide is removed as it passes into the cisternae of the rough endoplasmic reticulum, and a hexapeptide at the resulting amino terminal is subsequently cleaved off farther along the secretory pathway (see Figure 46–12). The synthesis of albumin is depressed in a variety of diseases, particularly those of the liver. The plasma of patients with liver disease often shows a decrease in the ratio of albumin to globulins (decreased albumin-globulin ratio). The synthesis of albumin decreases relatively early in conditions of protein malnutrition, such as kwashiorkor.
Mature human albumin consists of one polypeptide chain of 585 amino acids and contains 17 disulfide bonds. By the use of proteases, albumin can be subdivided into three domains, which have different functions. Albumin has an ellipsoidal shape, which means that it does not increase the viscosity of the plasma as much as an elongated molecule such as fibrinogen does. Because of its relatively low molecular mass (about 69 kDa) and high concentration, albumin is thought to be responsible for 75-80% of the osmotic pressure of human plasma. Electrophoretic studies have shown that the plasma of certain humans lacks albumin. These subjects are said to exhibit analbuminemia.One cause of this condition is a mutation that affects splicing. Subjects with analbuminemia show only moderate edema, despite the fact that albumin is the major determinant of plasma osmotic pressure. It is thought that the amounts of the other plasma proteins increase and compensate for the lack of albumin.
Another important function of albumin is its ability to bind various ligands. These include free fatty acids (FFA), calcium, certain steroid hormones, bilirubin, and some of the plasma tryptophan. In addition, albumin appears to play an important role in transport of copper in the human body (see below). A variety of drugs, including sulfonamides, penicillin G, dicumarol, and aspirin, are bound to albumin; this finding has important pharmacologic implications.
Preparations of human albumin have been widely used in the treatment of hemorrhagic shock and of burns. However, some recent studies question the value of this therapy.
Haptoglobin Binds Extracorpuscular Hemoglobin, Preventing Free Hemoglobin from Entering the Kidney
Haptoglobin (Hp) is a plasma glycoprotein that binds extracorpuscular hemoglobin (Hb) in a tight noncovalent complex (Hb-Hp). The amount of haptoglobin in human plasma ranges from 40 to 180 mg of hemoglobin-binding capacity per deciliter. Approximately 10% of the hemoglobin that is degraded each day is released into the circulation and is thus extracor-puscular. The other 90% is present in old, damaged red blood cells, which are degraded by cells of the histiocytic system. The molecular mass of hemoglobin is approximately 65 kDa, whereas the molecular mass of the simplest polymorphic form of haptoglobin (Hp 1-1) found in humans is approximately 90 kDa. Thus, the Hb-Hp complex has a molecular mass of approximately 155 kDa. Free hemoglobin passes through the glomerulus of the kidney, enters the tubules, and tends to precipitate therein (as can happen after a massive incompatible blood transfusion, when the capacity of haptoglobin to bind hemoglobin is grossly exceeded) (Figure 50–3). However, the Hb-Hp complex is too large to pass through the glomerulus. The function of Hp thus appears to prevent loss of free hemoglobin into the kidney. This conserves the valuable iron present in hemoglobin, which would otherwise be lost to the body.

FIGURE 50–3 Different fates of free hemoglobin and of the hemoglobin-haptoglobin complex.
Human haptoglobin exists in three polymorphic forms, known as Hp 1-1, Hp 2-1, and Hp 2-2. Hp 1-1 migrates in starch gel electrophoresis as a single band, whereas Hp 2-1 and Hp 2-2 exhibit much more complex band patterns. Two genes, designated Hp1 and Hp2, direct these three phenotypes, with Hp 2-1 being the heterozygous phenotype. It has been suggested that the haptoglobin polymorphism may be associated with the prevalence of many inflammatory diseases.
The levels of haptoglobin in human plasma vary and are of some diagnostic use. Low levels of haptoglobin are found in patients with hemolytic anemias. This is explained by the fact that whereas the half-life of haptoglobin is approximately 5 days, the half-life of the Hb-Hp complex is about 90 min, the complex being rapidly removed from plasma by hepatocytes. Thus, when haptoglobin is bound to hemoglobin, it is cleared from the plasma about 80 times faster than normally. Accordingly, the level of haptoglobin falls rapidly in situations where hemoglobin is constantly being released from red blood cells, such as occurs in hemolytic anemias. Haptoglobin is an acute phase protein, and its plasma level is elevated in a variety of inflammatory states.
Haptoglobin-related protein is another protein found in human plasma. It bears a high degree of homology to haptoglobin and it appears to bind hemoglobin. Its level is elevated in some patients with cancers, although the significance of this is not understood.
Certain other plasma proteins bind heme but not hemoglobin. Hemopexin is a β1-globulin that binds free heme. Albumin will bind some metheme (ferric heme) to form methemalbumin, which then transfers the metheme to hemopexin.
IRON IS A VERY IMPORTANT BODY CONSTITUENT AND IS ZEALOUSLY CONSERVED
Transferrin is an important plasma protein involved in iron transport. Before discussing it and a number of other proteins involved in iron homeostasis, we shall describe certain aspects of iron and its metabolism.
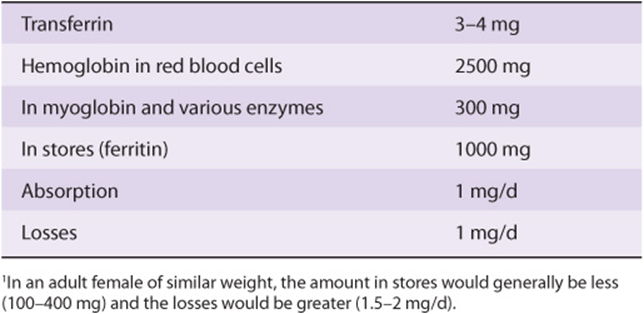
Iron is important in the human body because of its occurrence in many hemoproteins such as hemoglobin, myoglobin, and the cytochromes (including the cytochrome P450 group of enzymes). A normal 70 kg adult male has 3-4 g of iron in his body, the distribution of which is shown in Table 50-3. Under normal circumstances, the body guards its content of iron zealously. A healthy adult male loses only about 1 mg/day, which is replaced by absorption from the intestine. Adult females (premenopausal) require about 1.5 mg/day and are more prone to develop states of iron deficiency because of blood loss during menstruation.
TABLE 50–3 Distribution of Iron in a 70-kg Adult Male1
Absorption of Dietary Iron Occurs in the Duodenum
Iron is ingested in the diet either as non-heme or heme iron. Absorption of iron by enterocytes of the proximal duodenum is a highly regulated process (Figure 50–4). Inorganic dietary iron in the ferric state (Fe3+) is reduced to its ferrous form (Fe2+) by a brush border membrane-bound ferrireductase, duodenal cytochrome b (Dcytb). Vitamin C, gastric acid, and a number of other reducing agents present in food may also favor reduction of ferric to ferrous iron. The transfer of iron across the apical membrane of the enterocytes is accomplished via the divalent metal transporter 1 (DMT1 or SLC11A2). DMT1 is relatively non-specific and may also be involved in the transport of other divalent cations, such as Mn2+, Co2+, Zn2+, Cu2+, and Pb2+. Once inside the enterocytes, iron can either be stored as ferritin or transferred across the basolateral membrane into the circulation by the iron exporter protein, ferroportin or iron-regulated protein 1 (IREG1 or SLC40A1). This process occurs in conjunction with hephaestin, a copper-containing ferroxidase homologous to ceruloplasmin, which oxidizes Fe2+ to Fe3+. Iron is transported in plasma in the Fe3+ form by the transport protein, transferrin. Excess iron that is stored in the enterocytes as ferritin is lost when the enterocytes are sloughed off into the gut lumen.
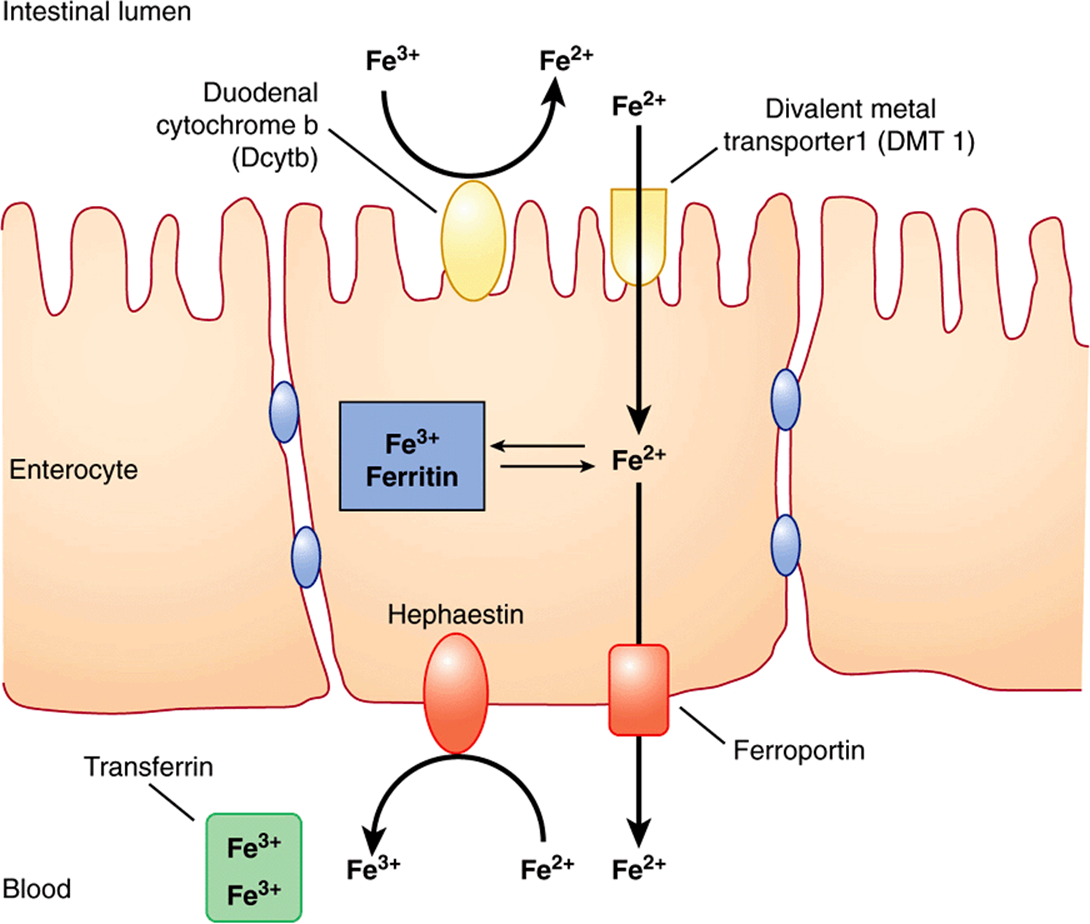
FIGURE 50–4 Non-heme iron transport in enterocytes. Ferric iron is reduced to the ferrous form by a luminal ferrireductase, duodenal cytochrome b (Dcytb). Ferrous iron is transported into the enterocyte via divalent metal transporter-1 (DMT-1). Within the enterocyte, iron is either stored as ferritin, or transported out of the cell, across the baslolateral membrane, by ferroportin (Fp). Ferrous iron is oxidized to its ferric form by the ferroxidase, hephaestin. Ferric iron is then bound by transferrin in blood which transports it to various sites in the body. (Based on Andrews NC: Forging a field: the golden age of iron biology. Blood 2008;112(2);219.)
Heme iron in the diet is taken up by enterocytes by mechanisms independent of those involved in the uptake of dietary inorganic iron. Heme in enterocytes is broken down by heme oxygenase (HO, see Chapter 31) to release iron, which is either stored as ferritin or transported into circulation by ferroportin.
Transferrin Shuttles Iron to Sites Where It Is Needed
Free iron is extremely toxic because of its ability to catalyze the formation of harmful oxygen free radicals, via the Fenton reaction (Figure 50–5). In biological systems, iron is always bound to proteins in order to limit the generation of toxic radicals. In the plasma, iron is tightly bound to the plasma protein, transferrin (Tf), which plays a central role in transporting iron around the body to sites where it is needed. It is a β1 - globulin with a molecular mass of approximately 76 kDa. It is a glycoprotein and is synthesized in the liver. It transports iron in circulation to sites where iron is required (eg, the bone marrow). It has two high-affinity binding sites for Fe3+ iron. Transferrin, when bound to two atoms of iron, is called holotransferrin (Tf-Fe). The concentration of Tf in plasma is approximately 300 mg/dL. This amount of transferrin can bind a total of approximately 300 μg of iron (per deciliter of plasma). This represents the total iron-binding capacity (TIBC) of plasma. Normally, transferrin is about 30% saturated with iron. Transferrin saturation decreases to less than 16% during severe iron deficiency and may increase to more than 45% in iron overload conditions.
![]()
FIGURE 50–5 The Fenton reaction. Free iron is extremely toxic as it can catalyze the formation of hydroxyl radical (OH–) from hydrogen peroxide (see also Chapter 52). The hydroxyl radical is a transient but highly reactive species and can oxidize cellular macromolecules resulting in tissue damage.
Glycosylation of transferrin is impaired in congenital disorders of glycosylation (Chapter 47) and chronic alcoholism, resulting in increased circulating levels of carbohydrate-deficient transferrin (CDT). CDT can be measured by isoelectric focussing (IEF) and is used as a marker of chronic alcoholism.
The Transferrin Cycle Helps in Cellular Uptake of Iron
Transferrin receptor 1 (TfR1) is present on the surface of almost all cells, especially erythroid precursors in the bone marrow. Transferrin binds to these receptors and is internalized by receptor-mediated endocytosis (similar to LDL receptors described in Chapter 25). The acidic pH inside the late endosome causes iron to dissociate from transferrin (Tf). The dissociated iron leaves the endosome via DMT1 to enter the cytoplasm. Unlike the protein component of LDL, apoTf (Tf without iron bound to it) is not degraded within the endosome. Instead, it remains associated with its receptor and returns to the plasma membrane. It can then dissociate from its receptor, re-enter plasma, and pick up more iron to be delivered to cells. This is called the transferrin cycle (Figure 50–6).
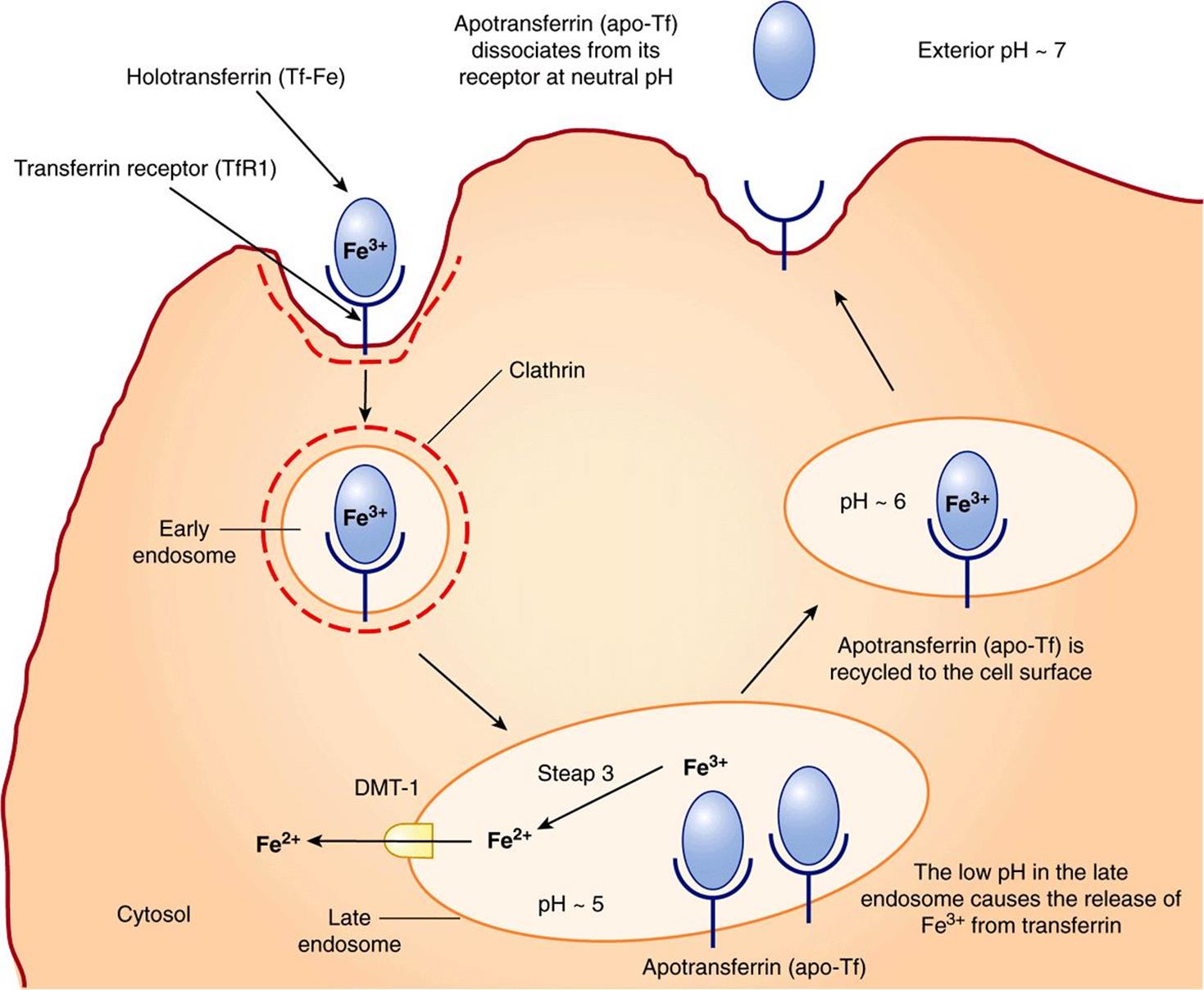
FIGURE 50–6 The transferrin cycle. Holotransferrin (Tf-Fe) binds to transferrin receptor 1 (TfR1) present in clathrin-coated pits on the cell surface. The TfR1-Tf-Fe complex is endocytosed and endocytic vesicles fuse to form early endosomes. The early endosomes mature to late endosomes, which have an acidic pH inside. The low pH causes release of iron from its binding sites on transferrin. Apotransferrin (apo-Tf) remains bound to TfR1. Ferric iron is converted to its ferrous form by the ferrireductase, Steap 3. Ferrous iron is then transported into the cytosol via DMT1. The TfR1-apo-Tf complex is recycled back to the cell surface. At the cell surface, apo-Tf is released from TFR1. TfR1 then binds to new Tf-Fe. This completes the transferrin cycle. (Based on Figure 17–48 in Lodish H et al: Molecular Cell Biology, 4th ed. WH Freeman, 2000).
Transferrin receptor 2 (TfR2) is expressed primarily on the surface of hepatocytes and also in the crypt cells of the small intestine. It has a low affinity for Tf-Fe and does not appear to be involved in iron uptake by cells. It plays a role in sensing body iron stores in association with other proteins, as discussed later.
Iron in Senescent Erythrocytes Is Recycled by Macrophages
Normally erythrocytes have a lifespan of approximately 120 days. Senescent or damaged erythrocytes are phagocytosed by macrophages of the reticuloendothelial system (RES) present in the spleen and liver. Around 200 billion erythrocytes (in about 40 mL of blood) are catabolized every day in this way. Within the macrophage, heme derived from hemoglobin is broken down by heme oxygenase, converting it to biliverdin. Carbon monoxide and iron are released as by-products. Iron released from heme is exported from the phagocytic vesicle in the macrophage by NRAMP 1 (natural resistance-associated macrophage protein 1), a transporter homologous to DMT1. It is subsequently transported into the circulation via ferroportin in the plasma membrane of the macrophage (Figure 50–7). Therefore, ferroportin plays a central role, not only in iron absorption in the intestine, but also in iron release from macrophages. Ceruloplasmin (see below) is a copper-containing plasma protein synthesized by liver. It has ferrioxidase activity. Ceruloplasmin is required for the oxidation of Fe2+ to Fe3+. Fe3+ is then bound to transferrin in blood. The iron released from macrophages in this way (about 25 mg per day) is recycled and forms the major source of iron for the body. In comparison, intestinal iron absorption contributes only 1-2 mg of the body’s daily iron needs.
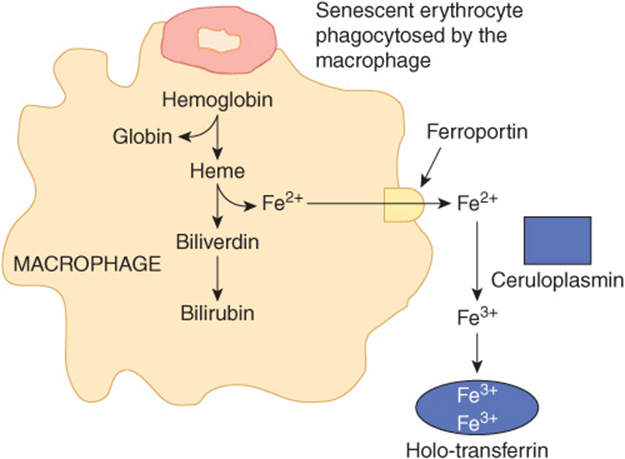
FIGURE 50–7 Recycling of iron in macrophages. Senescent erythrocytes are phagocytosed by macrophages. Hemoglobin is degraded and iron is released from heme by the action of the enzyme heme oxygenase. Iron, in the ferrous form, is then transported out of the macrophage via ferroportin (Fp). In the plasma, it is oxidized to the ferric form by ceruloplasmin before binding to transferrin (Tf). Iron circulates in blood tightly bound to Tf.
Ferritin Stores Iron in Cells
Under normal circumstances, ferritin stores excess iron in various tissues and constitutes approximately 1 g of the total body iron content. Ferritin has a molecular mass of approximately 440 kDa. It is composed of 24 subunits, which surround 3000-4500 ferric atoms. The subunits may be of the H (heavy) or the L (light) type. The H-subunit possesses ferroxidase activity which is required for iron-loading of ferritin. The function of the L subunit is not clearly known but is proposed to play a role in ferritin nucleation and stability. Normally, there is a small amount of ferritin in human plasma (50-200 μg/dL) proportionate to the total stores of iron in the body. Plasma ferritin levels are, thus, considered to be an indicator of body iron stores. However, it is not known whether ferritin in plasma is derived from damaged cells or actively secreted by cells.
Hemosiderin is an ill-defined molecule and appears to be a partly degraded form of ferritin that contains iron. It can be detected in tissues, under conditions of iron overload (hemosiderosis), by histological stains (eg, Prussian blue).
Intracellular Iron Homeostasis Is Tightly Regulated
Syntheses of TfR1 and ferritin are reciprocally linked to intracellular iron content. When iron levels are high, ferritin is synthesized to store iron and, since no further uptake of iron is required, synthesis of TfR1 is inhibited. Conversely, when iron levels are low, ferritin is not synthesized while TfR1 is, in order to promote uptake of iron from transferrin in blood.
The mechanisms involved in the regulation of syntheses of ferritin and TfR1 have been elucidated (Figure 50–8). This is brought about through regulation of the stability of the mRNAs for ferritin and TfR1. The ferritin and TfR1 mRNAs contain iron response elements (IREs), which form hairpin loops at their 5′ and 3′ untranslated regions (UTRs), respectively. IREs are bound by iron regulatory proteins (IRPs). The IRPs are sensitive to intracellular iron levels and are induced by low levels. They bind IREs only when intracellular iron levels are low. Binding of IRP to the IRE at the 3′ UTR of TfR1 mRNA stabilizes the TfR1 mRNA, thus increasing TfR1 synthesis and expression on the cell surface. On the other hand, binding of IRP to the IRE at the 5′ UTR of ferritin mRNA blocks translation of ferritin. Similarly, in the absence of IRP binding to IRE (which happens in the presence of high levels of iron), translation of ferritin mRNA is facilitated and TfR1 mRNA is rapidly degraded. The net result is that, when intracellular iron levels are high, ferritin is synthesized but TfR1 is not, and when intracellular iron levels are low, TfR1 is synthesized and ferritin is not. This is a classical example of control of expression of proteins at the translational level.
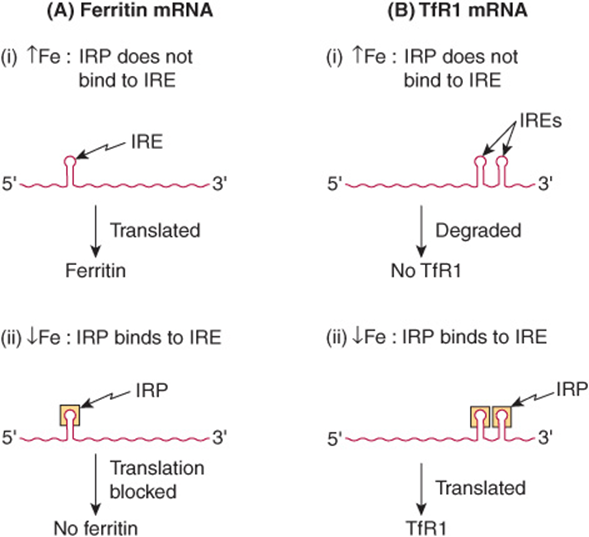
FIGURE 50–8 Schematic representation of the reciprocal relationship between synthesis of ferritin and the transferrin receptor (TfR1). The mRNA for ferritin is represented on the left, and that for TfR1 on the right of the diagram. At high concentrations of iron, the iron bound to the IRP prevents that protein from binding the IREs on either type of mRNA. The mRNA for ferritin is able to be translated under these circumstances, and ferritin is synthesized. On the other hand, when the IRP is not able to bind to the IRE on the mRNA for TfR1, that mRNA is degraded. In contrast, at low concentrations of iron, the IRP is able to bind to the IREs on both types of mRNA. In the case of the ferritin mRNA, this prevents it from being translated. Hence ferritin is not synthesized. In the case of the mRNA for TfR1, binding of the IRP prevents that mRNA from being degraded, it is translated, and TfR1 is synthesized. IRP, iron regulatory protein; IRE, iron response element.
Hepcidin Is the Chief Regulator of Systemic Iron Homeostasis
Hepcidin is a protein that is known to play a central role in iron homeostasis in the body. It is synthesized by the liver as an 84-aminoacid precursor protein (prohepcidin). Prohepcidin is cleaved to generate bioactive hepcidin, which is a 25-amino-acid peptide. Hepcidin binds to the cellular iron exporter, ferroportin, and triggers its internalization and degradation. Thus, as shown in Figure 50–9, hepcidin decreases iron absorption in the intestine (producing a “mucosal block”) and also prevents recycling of iron from macrophages. These effects result in a reduction in circulating iron levels (hypoferremia). In addition, it reduces placental iron transfer (not shown in Figure 50–9). When plasma iron levels are high, hepatic synthesis of hepcidin increases, thus reducing iron absorption and macrophage iron recycling. The opposite occurs when plasma iron levels are low.
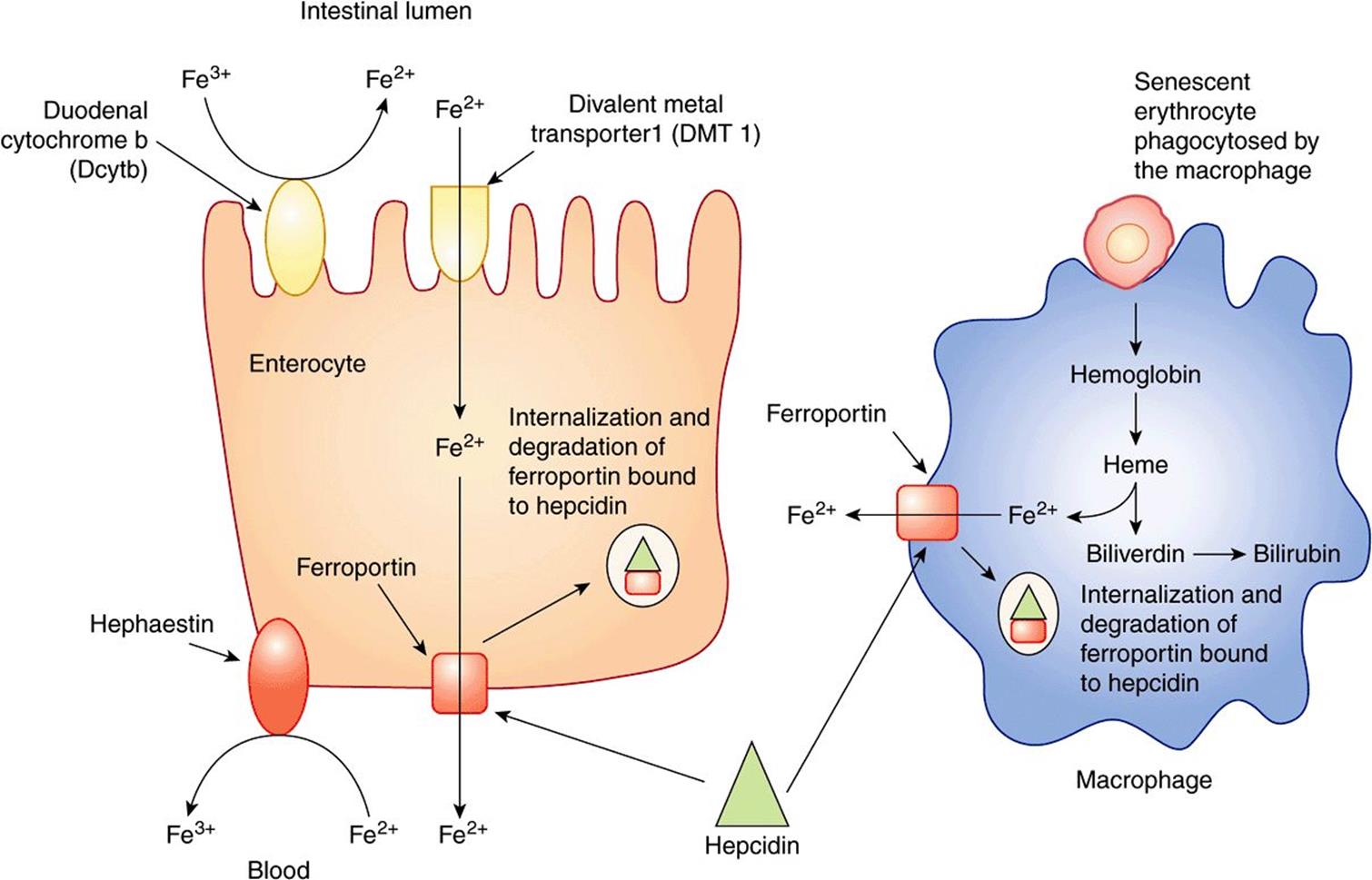
FIGURE 50–9 Role of hepcidin in systemic iron regulation. Hepcidin binds to and triggers the internalization and degradation of ferroportin expressed on the surface of enterocytes and macrophages. This decreases iron absorption from the intestine and inhibits iron release from macrophages, leading to hypoferremia. (Based on Andrews NC: Forging a field: the golden age of iron biology. Blood 2008;112(2): 219.)
The expression of hepcidin in the liver is a highly regulated process, many aspects of which are still under investigation. Hepcidin expression is regulated by systemic iron availability, erythropoiesis, inflammation, and hypoxia, among other signals. Studies on patients with hereditary hemochromatosis (described below and Case 10 in Chapter 57) have yielded a large amount of information regarding hepcidin regulation.
Hepatocytes have an “iron-sensing complex” on their surface. There are several proteins that constitute this complex (see Figure 50–10). These include the HFE protein (which is most commonly mutated in hereditary hemochromatosis), TfR1, TfR2, and hemojuvelin (HJV). The HFE protein is a major histocompatibility (MHC) class 1-like molecule that is expressed on the cell surface, bound to β2-microglobulin (a component of class I MHC molecules, not shown in Figure 50–10) and TfR1. HFE binds to TfR1 at a site that overlaps with its binding site for holotransferrin (Tf-Fe). Tf-Fe, therefore, competes with HFE for binding to TfR1. When displaced from TfR1 by Tf-Fe (which happens when Tf-Fe levels are high), HFE binds to TfR2, which is also expressed on the surface of hepatocytes. The HFE-TfR2 complex is further stabilized by binding to Tf-Fe. This complex triggers an intracellular signaling cascade which ultimately results in upregulation of expression of the hepcidin gene (HAMP). The critical roles of HFE, HJV, and TfR2 in hepcidin regulation are demonstrated by the fact that mutations in their genes (like mutations in HAMP) are all characterized by low circulating levels of hepcidin and iron overload.
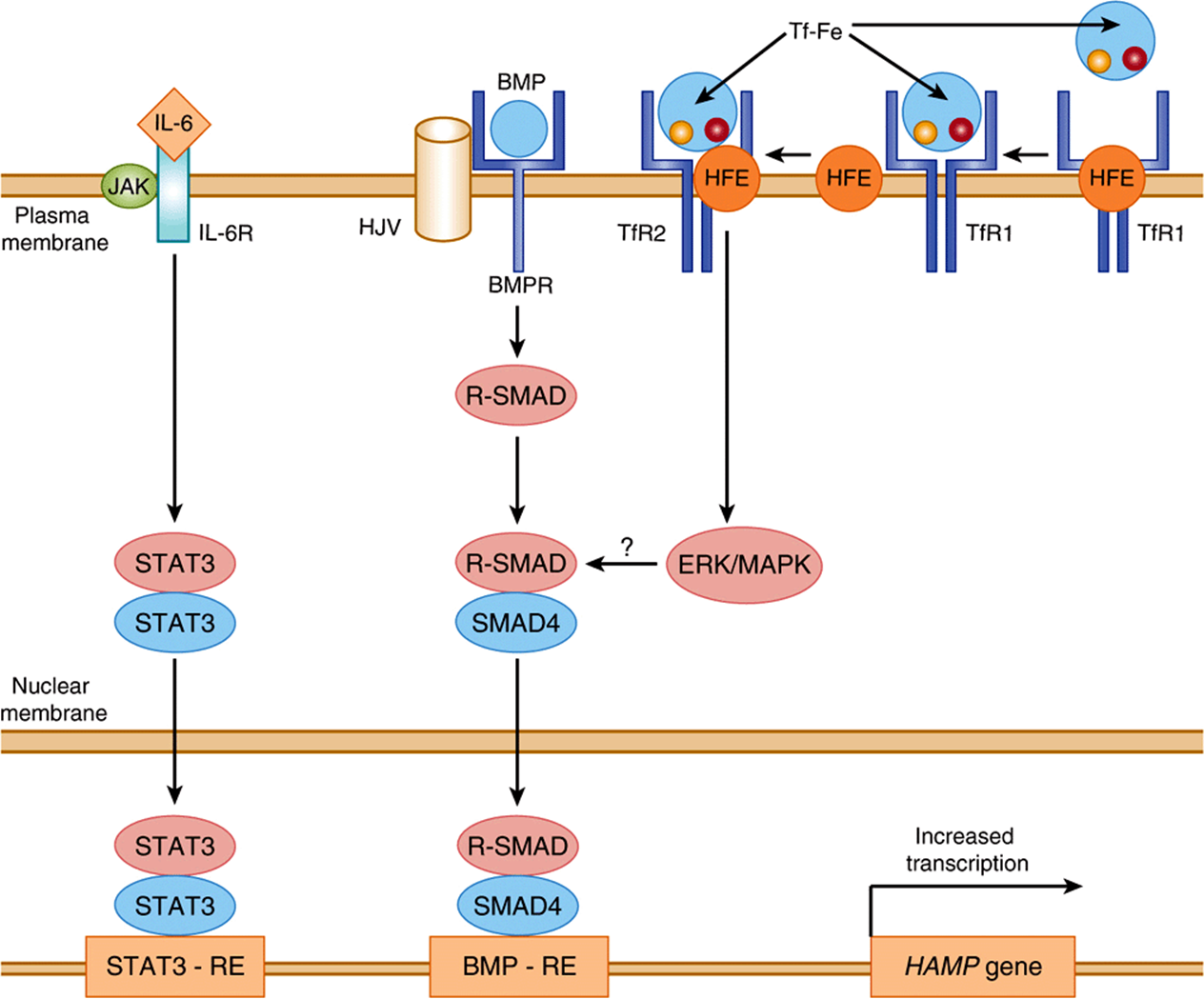
FIGURE 50–10 Regulation of hepcidin gene expression. Tf-Fe (holotransferrin) competes with HFE for binding to TFR1. High levels of Tf-Fe displace HFE from its binding site on TfR1. Displaced HFE binds to TfR2 along with Tf-Fe to signal via the ERK/MAPK pathway to induce hepcidin. BMP binds to its receptor BMPR and HJV (co-receptor) to activate R-SMAD. R-SMAD dimerizes with SMAD4, translocates to the nucleus where it binds to the BMP-RE, resulting in transcriptional activation of hepcidin as shown. IL-6, which is a biomarker of inflammation, binds to its cell-surface receptor and activates the JAK-STAT pathway. STAT3 translocates to the nucleus where it binds to its response element (STAT-RE) on the hepcidin gene to induce it. BMP-RE, BMP response element; BMP, bone morphogenetic protein; BMPR, bone morphogenetic protein receptor; ERK-MAPK, extracellular signal-regulated kinase/mitogen-activated protein kinase; HAMP, gene encoding hepcidin antimicrobial peptide (hepcidin); HJV, hemojuvelin; IL-6, interleukin 6; IL-6R, interleukin 6 receptor; JAK, Janus-associated kinase; SMAD, Sma and MAD (Mothers Against Decapentaplegic)-related protein; STAT, signal transduction and activator of transcription; STAT3-RE, STAT 3 response element; TfR1, transferrin receptor 1; TfR2, transferrin receptor 2. (Redrawn from Hentz MW, Muckenthaler MU, Gali B et al: Two to tango: regulation of mammalian iron metabolism. Cell 2010;142:24.)
Bone morphogenetic proteins (BMPs), especially BMP6, play an important role in regulating basal hepcidin expression. BMPs act by mechanisms that are distinct from HFE, but considerable cross-talk exists between these pathways. BMP levels are regulated by iron by mechanisms that have not yet been fully elucidated. BMP binds to its cell-surface receptors (BMPR). This binding is facilitated by HJV, which acts as a BMP co-receptor. The activation of the BMPR-HJV complex causes phosphorylation of SMAD (intracellular signaling proteins) (Figure 50–10), which subsequently results in transcriptional activation of hepcidin.
Hepcidin levels are also regulated by erythropoietic signals. For example, it is known that hepcidin levels are down-regulated in β-thalassemia major, which is characterized by ineffective erythropoiesis and iron overload. Recently, growth differentiation factor 15 (GDF15) and twisted gastrulation 1 (TWSG1) have been shown to be critical molecules secreted by erythroblasts. These downregulate hepatic hepcidin expression in β-thalassemia.
Inflammation is known to induce hepcidin expression. Interleukin-6 (IL-6), an inflammatory cytokine, signals through the JAK-STAT (Janus Kinase—Signal Transducer and Activator of Transcription) pathway to mediate this effect (Figure 50–10). Anemia that is associated with chronic inflammation (anemia of inflammation or AI) is probably due to inflammation-mediated upregulation of hepcidin. AI manifests as a microcytic, hypochromic anemia that is refractory to iron supplementation. In addition to the above-mentioned factors, hypoxia is also known to induce hepcidin and this effect is mediated by stabilization of the hypoxia-inducible factors 1 and 2 (HIF-1 and HIF-2).
Iron Deficiency Is Highly Prevalent
Iron deficiency is extremely common in many parts of the world, especially in developing countries. There are several causes for iron deficiency. Decreased intake of iron in the diet and malabsorption are the most common causes in developing countries. Premenopausal women are more prone to developing iron deficiency since they have higher daily requirements of iron, owing to loss of blood during menstruation. Chronic blood losses, due to gastrointestinal bleeding or excessive menstruation, are also common causes for iron deficiency.
The development of iron deficiency anemia progresses through three stages—negative iron balance, iron-deficient erythropoiesis, and finally, iron-deficiency anemia. Negative iron balance is the initial stage where intestinal iron absorption is insufficient to meet the body’s demands. This results in the body’s iron stores being mobilized to meet requirements. This leads to progressive depletion of the iron stores. At this stage, all laboratory tests are normal, except for a low serum ferritin, which, as described above, is a marker of body iron stores. If this situation persists, iron stores are depleted and serum ferritin levels fall below 15 μg/dL. At this stage, the transferrin levels in blood increase resulting in an increase in the TIBC. The transferrin saturation, however, decreases and may fall below 20%. Hemoglobin synthesis is impaired. This is the stage of iron-deficient erythropoiesis. If iron deficiency is not corrected at this stage, hemoglobin levels in blood gradually begin to fall, resulting in the final stage of iron-deficiency anemia. This stage is characterized by a hypochromic, microcytic blood picture. Patients typically present with fatigue, pallor, and reduced exercise capacity. Table 50-4 gives a summary of changes in common laboratory tests at various stages in the development of iron deficiency anemia.
TABLE 50–4 Changes in Various Laboratory Tests Used to Assess Iron-Deficiency Anemia
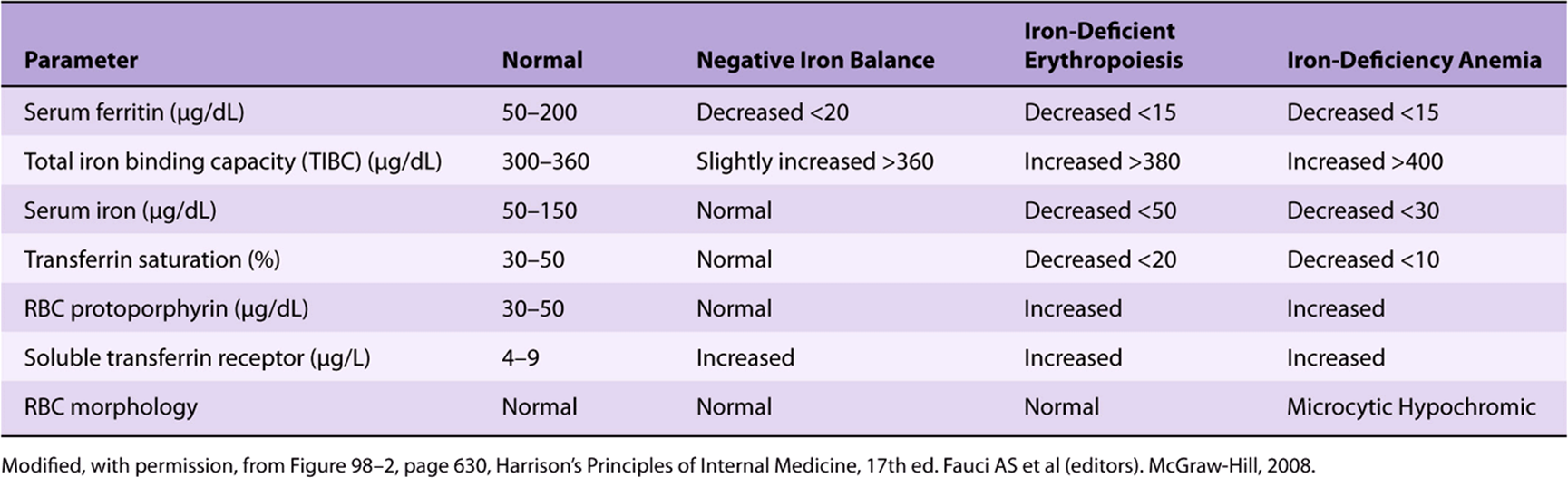
Red-cell protoporphyrin levels are increased in iron deficiency. The presence of protoporphyrin in erythrocytes reflects impaired ferrochelatase-catalyzed incorporation of iron into the protoporphyrin IX ring. Serum transferrin receptor protein or soluble transferrin receptor (sTfR) is also considered to be a useful marker of iron deficiency. Under normal conditions, TfR1 is highly expressed on the surface of erythroid cells and a certain proportion is released into the circulation by proteolytic cleavage. This is referred to as sTfR. Increased serum sTfR levels reflect increased expression of TfR1 on the erythroid cell surface during iron deficiency. Estimation of the serum sTfR level is especially useful in distinguishing between anemia due to iron deficiency and that due to chronic inflammation (AI, described above). It is elevated in the former and remains within the reference range in the later condition. Serum ferritin is not useful in this situation because, ferritin, being an acute phase protein, increases during inflammation, even in the presence of anemia.
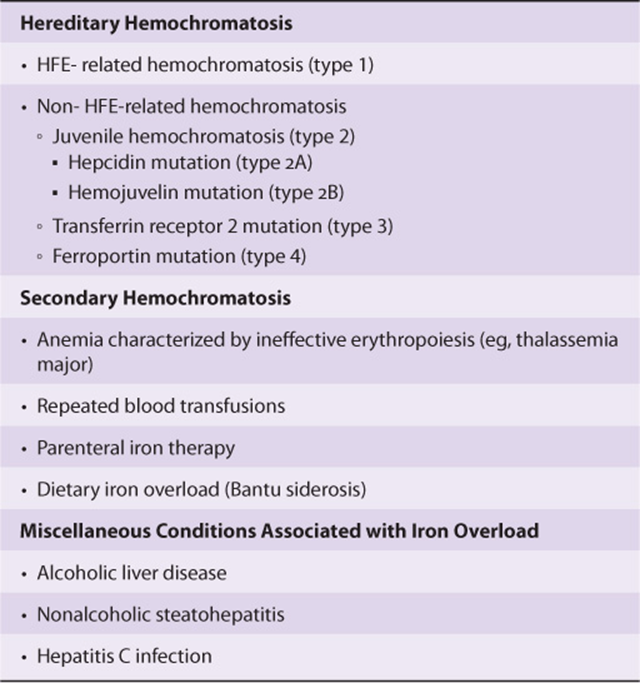
Hereditary Hemochromatosis Is Characterized by Iron Overload
Iron overload conditions, also called hemochromatosis, are characterized by increased intestinal iron absorption resulting in increased total body iron stores. The term hemosiderosis is used to signify the presence of stainable iron in tissues, which is often a characteristic feature of iron overload conditions.
Iron overload may be hereditary or secondary (Table 50-5). Hereditary hemochromatosis most often is caused by mutations in the HFE gene. Rarer forms of this disease may arise from mutations in the hepcidin (HAMP),TfR2, HJV, and ferroportin genes. Secondary iron overload is usually associated with ineffective erythropoiesis, as seen in the thalassemia syndromes. Repeated blood transfusions can also result in progressive iron overload. Additional details of the causes, pathogenesis, and clinical manifestations of hereditary hemochromatosis are discussed under Case 10, Chapter 57.
TABLE 50–5 Iron Overload Conditions
Ceruloplasmin Binds Copper, & Low Levels of This Plasma Protein Are Associated With Wilson Disease
Ceruloplasmin (about 160 kDa) is an a2-globulin. It has a blue color because of its high copper content and carries 90% of the copper present in plasma. Each molecule of ceruloplasmin binds six atoms of copper very tightly, so that the copper is not readily exchangeable. Albumin carries the other ~10% of the plasma copper, but binds the metal less tightly than does ceruloplasmin. Albumin thus donates its copper to tissues more readily than ceruloplasmin and appears to be more important than ceruloplasmin in copper transport in the human body. Ceruloplasmin exhibits a copper-dependent oxidase activity, but its physiologic significance has not been clarified apart from possible involvement in the oxidation of Fe2+ in transferrin to Fe3+ (see above). The amount of ceruloplasmin in plasma is decreased in liver disease. In particular, low levels of ceruloplasmin are found in Wilson disease (hepatolenticular degeneration), a disease due to abnormal metabolism of copper. In order to clarify the description of Wilson disease, we shall first consider the metabolism of copper in the human body and then Menkes disease, another condition involving abnormal copper metabolism.
Copper Is a Cofactor for Certain Enzymes
Copper is an essential trace element. It is required in the diet because it is the metal cofactor for a variety of enzymes (see Table 50-6). Copper plays important roles in cellular respiration (cytochrome c oxidase), iron homeostasis (ceruloplasmin), melanin formation (tyrosinase), neurotransmitter production (various enzymes), synthesis of connective tissue (lysyl oxidase), and protection against oxidants (eg, superoxide dismutase). It accepts and donates electrons and is involved in reactions involving dismutation, hydroxylation, and oxygenation. However, excess copper can cause problems because it can oxidize proteins and lipids, bind to nucleic acids, and enhance the production of free radicals. It is thus important to have mechanisms that will maintain the amount of copper in the body within normal limits. The body of the normal adult contains about 100 mg of copper, located mostly in bone, liver, kidney, and muscle. The daily intake of copper is about 2-4 mg, with about 50% being absorbed in the stomach and upper small intestine and the remainder excreted in the feces. Copper is carried to the liver bound to albumin, taken up by liver cells, and part of it is excreted in the bile. Copper also leaves the liver attached to ceruloplasmin, which is synthesized in that organ.
TABLE 50–6 Some Important Enzymes That Contain Copper

The Tissue Levels of Copper & of Certain Other Metals Are Regulated in Part by Metallothioneins
Metallothioneins are a group of small proteins (about 6.5 kDa), found in the cytosol of cells, particularly of liver, kidney, and intestine. They have a high content of cysteine and can bind copper, zinc, cadmium, and mercury. The SH groups of cysteine are involved in binding the metals. Acute intake (eg, by injection) of copper and of certain other metals increases the amount (induction) of these proteins in tissues, as does administration of certain hormones or cytokines. These proteins may function to store the above metals in a nontoxic form and are involved in their overall metabolism in the body. Sequestration of copper also diminishes the amount of this metal available to generate free radicals.
Menkes Disease Is Due to Mutations in the Gene Encoding a Copper-Binding P-Type ATPase
Menkes disease (“kinky” or “steely” hair disease) is a disorder of copper metabolism. It is X-linked, affects only male infants, involves the nervous system, connective tissue, and vasculature, and is usually fatal in infancy. Early diagnosis is important, because injections of copper may be effective if the condition is treated promptly. In 1993, it was reported that the basis of Menkes disease was mutations in the gene (the ATP7A gene) for a copper-binding P-type ATPase (the ATP7A protein). Interestingly, the enzyme showed structural similarity to certain metal-binding proteins in microorganisms. This ATPase is thought to be responsible for directing the efflux of copper from cells. When altered by mutation, copper is not mobilized normally from the intestine, in which it accumulates, as it does in a variety of other cells and tissues, from which it cannot exit. Despite the accumulation of copper, the activities of many copper-dependent enzymes are decreased, perhaps because of a defect of its incorporation into the apoenzymes. Normal liver expresses very little of the ATPase, which explains the absence of hepatic involvement in Menkes disease. This work led to the suggestion that liver might contain a different copper-binding ATPase, which could be involved in the causation of Wilson disease. As described below, this turned out to be the case.
Wilson Disease Is Also Due to Mutations in a Gene Encoding a Copper-Binding P-Type ATPase
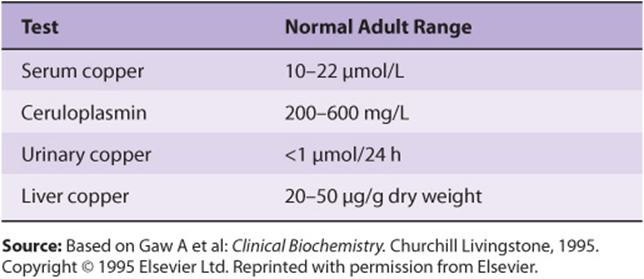
Wilson disease is a genetic disease in which copper fails to be excreted in the bile and accumulates in liver, brain, kidney, and red blood cells. It can be regarded as an inability to maintain a near-zero copper balance, resulting in copper toxicosis. The increase of copper in liver cells appears to inhibit the coupling of copper to apoceruloplasmin and leads to low levels of ceruloplasmin in plasma. As the amount of copper accumulates, patients may develop a hemolytic anemia, chronic liver disease (cirrhosis and hepatitis), and a neurologic syndrome owing to accumulation of copper in the basal ganglia and other centers. A frequent clinical finding is the Kayser-Fleischer ring. This is a green or golden pigment ring around the cornea due to deposition of copper in Descemet’s membrane. The major laboratory tests of copper metabolism are listed in Table 50-7. If Wilson disease is suspected, a liver biopsy should be performed; a value for liver copper of over 250 μg/g dry weight along with a plasma level of ceruloplasmin of under 20 mg/dL is diagnostic.
TABLE 50–7 Major Laboratory Tests Used in the Investigation of Diseases of Copper Metabolism
The cause of Wilson disease was also revealed in 1993, when it was reported that a variety of mutations in a gene encoding a copper-binding P-type ATPase (ATP7B protein) were responsible. The gene (ATP7B) is estimated to encode a protein of 1411 amino acids, which is highly homologous to the product of the gene affected in Menkes disease. In a manner not yet fully explained, a nonfunctional ATPase causes defective excretion of copper into the bile, a reduction of incorporation of copper into apoceruloplasmin, and the accumulation of copper in liver and subsequently in other organs such as brain.
Treatment for Wilson disease consists of a diet low in copper along with lifelong administration of penicillamine, which chelates copper, is excreted in the urine, and thus depletes the body of the excess of this mineral.
Another condition involving ceruloplasmin is aceruloplasminemia. In this genetic disorder, levels of ceruloplasmin are very low and consequently its ferroxidase activity is markedly deficient. This leads to failure of release of iron from cells, and iron accumulates in certain brain cells, hepatocytes, and pancreatic islet cells. Affected individuals show severe neurologic signs and have diabetes mellitus. Use of a chelating agent or administration of plasma or ceruloplasmin concentrate may be beneficial.
Genetic Deficiency of α1-Antiproteinase (α1-Antitrypsin) Is Associated With Emphysema & One Type Of Liver Disease
α1-Antiproteinase (about 52 kDa) was formerly called α1-antitrypsin, and this name is retained here. It is a single-chain protein of 394 amino acids, contains three oligosaccharide chains, and is the major component (>90%) of the α1, fraction of human plasma. It is synthesized by hepatocytes and macrophages and is the principal serine protease inhibitor (serpin, or Pi) of human plasma. It inhibits trypsin, elastase, and certain other proteases by forming complexes with them. At least 75 polymorphic forms occur, many of which can be separated by electrophoresis. The major genotype is MM, and its phenotypic product is PiM. There are two areas of clinical interest concerning α1,-antitrypsin. A deficiency of this protein has a role in certain cases (approximately 5%) of emphysema. This occurs mainly in subjects with the ZZ genotype, who synthesize PiZ, and also in PiSZ heterozygotes, both of whom secrete considerably less protein than PiMM individuals. Considerably less of this protein is secreted as compared with PiM. When the amount of α1-antitrypsin is deficient and polymorphonuclear white blood cells increase in the lung (eg, during pneumonia), the affected individual lacks a countercheck to proteolytic damage of the lung by proteases such as elastase (Figure 50–11). It is of considerable interest that a particular methionine (residue 358) of α-antitrypsin is involved in its binding to proteases. Smoking oxidizes this methionine to methionine sulfoxide and thus inactivates it. As a result, affected molecules of α1-antitrypsin no longer neutralize proteases. This is particularly devastating in patients (eg, PiZZ phenotype) who already have low levels of α1-antitrypsin. The further diminution in α1-antitrypsin brought about by smoking results in increased proteolytic destruction of lung tissue, accelerating the development of emphysema. Intravenous administration of α1-antitrypsin (augmentation therapy) has been used as an adjunct in the treatment of patients with emphysema due to α1-antitrypsin deficiency. Attempts are being made, using the techniques of protein engineering, to replace methionine 358 by another residue that would not be subject to oxidation. The resulting “mutant” α1-antitrypsin would thus afford protection against proteases for a much longer period of time than would native α1-antitrypsin. Attempts are also being made to develop gene therapy for this condition. One approach is to use a modified adenovirus (a pathogen of the respiratory tract) into which the gene for α1-antitrypsin has been inserted. The virus would then be introduced into the respiratory tract (eg, by an aerosol). The hope is that pulmonary epithelial cells would express the gene and secrete α1-antitrypsin locally. Experiments in animals have indicated the feasibility of this approach.
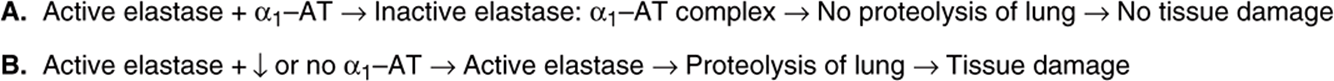
FIGURE 50–11 Scheme illustrating (A) normal inactivation of elastase by (α-antitrypsin and (B) situation in which the amount of (α1-antitrypsin is substantially reduced, resulting in proteolysis by elastase and leading to tissue damage.
Deficiency of α1-antitrypsin is also implicated in one type of liver disease α1-antitrypsin deficiency liver disease). In this condition, molecules of the ZZ phenotype accumulate and aggregate in the cisternae of the endoplasmic reticulum of hepatocytes. Aggregation is due to formation of polymers of mutant α1-antitrypsin, the polymers forming via a strong interaction between a specific loop in one molecule and a prominent β-pleated sheet in another (loop-sheet polymerization). By mechanisms that are not understood, hepatitis results with consequent cirrhosis (accumulation of massive amounts of collagen, resulting in fibrosis). It is possible that administration of a synthetic peptide resembling the loop sequence could inhibit loop-sheet polymerization. Diseases such as α1-antitrypsin deficiency, in which cellular pathology is primarily caused by the presence of aggregates of aberrant forms of individual proteins, have been named conformational diseases (see also Chapter 46). Most appear to be due to the formation of β-sheets by conformationally unstable proteins, which in turn leads to formation of aggregates. Other members of this group of conditions include Alzheimer disease, Parkinson disease, and Huntington disease.
At present, severe α1-antitrypsin deficiency liver disease can be successfully treated by liver transplantation. In the future, introduction of the gene for normal α1-antitrypsin into hepatocytes may become possible, but this would not stop production of the PiZ protein. Figure 50–12 is a scheme of the causation of this disease.
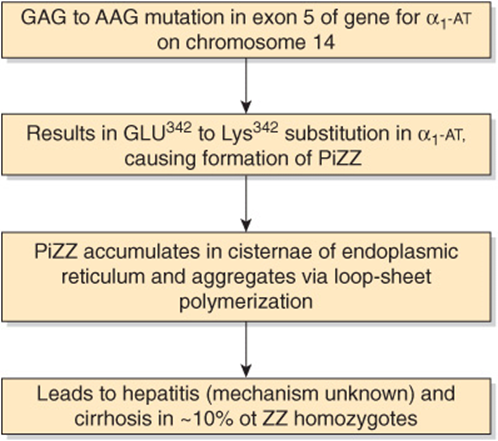
FIGURE 50–12 Scheme of causation of α1-antitrypsin-deficiency liver disease. The mutation shown causes formation of PiZZ (OMIM 107400). (α1-AT, α1-antitrypsin.)
α2-Macroglobulin Neutralizes Many Proteases & Targets Certain Cytokines to Tissues
α2-Macroglobulin is a large plasma glycoprotein (720 kDa) made up of four identical subunits of 180 kDa. It comprises 8-10% of the total plasma protein in humans. Approximately 10% of the zinc in plasma is transported by α2-macroglobulin, the remainder being transported by albumin. The protein is synthesized by a variety of cell types, including monocytes, hepatocytes, and astrocytes. It is the major member of a group of plasma proteins that include complement proteins C3 and C4. These proteins contain a unique internal cyclic thiol ester bond (formed between a cysteine and a glutamine residue, see Figure 50–13) and for this reason have been designated as the thiol ester plasma protein family. This bond is highly reactive and is involved in some of the biologic actions of α2-macroglobulin.
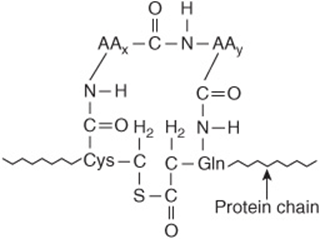
FIGURE 50–13 An internal cyclic thiol ester bond, as found in α2-macroglobulin. AAx and AAy are neighboring amino acids to cysteine and glutamine.
α2-Macroglobulin binds many proteinases and is thus an important panproteinase inhibitor. The α2-macroglobulinproteinase complexes are rapidly cleared from the plasma by a receptor located on many cell types. In addition, α2-macroglobulin binds many cytokines (platelet-derived growth factor, transforming growth factor-β, etc) and appears to be involved in targeting them toward particular tissues or cells. Once taken up by cells, the cytokines can dissociate from α2 macroglobulin and subsequently exert a variety of effects on cell growth and function. The binding of proteinases and cytokines by α2-macroglobulin involves different mechanisms that will not be considered here.
Amyloidosis Occurs by the Deposition of Proteins or Protein Fragments in Various Tissues
Amyloidosis is the accumulation of various insoluble fibrillar proteins between the cells of tissues to an extent that affects function. The accumulation is generally due to either increased production of certain proteins or accumulation of mutated forms of other proteins (see below). One or more organs or tissues may be affected, and the clinical picture depends on the sites and extent of deposition of amyloid fibrils. The fibrils generally represent proteolytic fragments of various plasma proteins and possess a β-pleated sheet structure. The term “amyloidosis” is a misnomer, as it was originally thought that the fibrils were starch-like in nature.
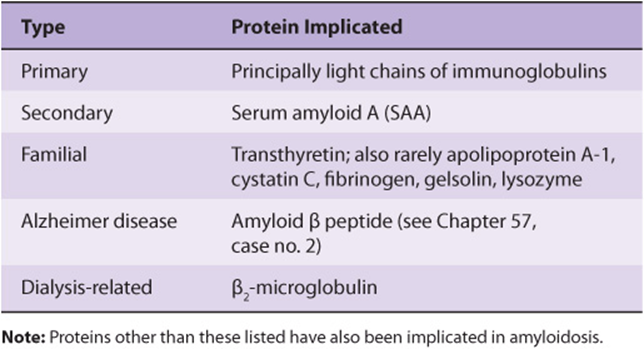
Amyloidosis is now generally classified as AX, where A represents amyloidosis and X the protein in the fibrils. However, this system will not be used here. A simple classification of amyloidosis is shown in Table 50-8. Primary amyloidosis is usually due to a monoclonal plasma cell disorder in which the protein that accumulates is a fragment of a light chain (see below) of an immunoglobulin. Secondary amyloidosis usually occurs secondary to chronic infections or cancer and is due to accumulation of degradation products of serum amyloid A (SAA). Increased synthesis of SAA occurs in chronic inflammatory states due to elevated levels of certain inflammatory cytokines that stimulate the liver to produce more of this protein. Familial amyloidosis results from accumulation of mutated forms of certain plasma proteins, particularly transthyretin (see Table 50-2). Over 80 mutated forms of this protein have been documented. Other plasma proteins can also accumulate in other rare types of familial amyloidosis. In patients undergoing long-term chronic dialysis, the plasma protein β2-microglobulin can accumulate, because it is retained in the plasma by dialysis membranes. Accumulation of an amyloid-type protein is believed to be a crucial factor in the causation of Alzheimer disease (see Case 2, Chapter 57). In all, at least 20 different proteins have been implicated in the different types of amyloidosis. The precise factors that determine the deposition of proteolytic fragments in tissues await elucidation. Amyloid fibrils generally have a P component associated with them, which is derived from serum amyloid P component, a plasma protein closely related to C-reactive protein. Tissue sections containing amyloid fibrils interact with Congo red dye and display striking green birefringence when viewed by polarizing microscopy. Deposition of amyloid occurs in patients with a variety of disorders; treatment of the underlying disorder should be provided if possible.
TABLE 50–8 A Classification of Amyloidosis
In general, experimental approaches to the treatment of amyloidosis can be considered under three headings: (1) preventing production of the precursor protein; (2) stabilizing the structures of precursor proteins so that they do not convert to β-pleated sheet structures; and (3) destabilizing amyloid fibrils so that they reconvert to their normal conformations. For instance, regarding the third approach, several small ligands bind avidly to amyloid fibrils. For example, iodinated anthracycline binds specifically and with high affinity to all natural amyloid fibrils and promotes their disaggregation in vitro. Another similar approach has been the development of the drug eprodisate.Amyloid fibrils bind to glycosaminoglycans (see Chapter 48) in tissues. Eprodisate binds to the GAGs, and thus disrupts the binding of the fibrils to these molecules. It is hoped that molecules affecting any of the three processes just mentioned may prove useful in the treatment of amyloidosis.
PLASMA IMMUNOGLOBULINS PLAY A MAJOR ROLE IN THE BODY’S DEFENSE MECHANISMS
The immune system of the body consists of three major components: B lymphocytes, T lymphocytes and the innate immune system. The B lymphocytes are mainly derived from bone marrow cells in higher animals and from the bursa of Fabricius in birds. The T lymphocytes are of thymic origin. The B cells are responsible for the synthesis of circulating, humoral antibodies, also known as immunoglobulins. The T cells are involved in a variety of important cell-mediated immunologic processes such as graft rejection, hypersensitivity reactions, and defense against malignant cells and many viruses. The innate immune system defends against infection in a nonspecific manner and unlike B and T cells is not adaptive. It contains a variety of cells such as phagocytes, neutrophils, natural killer cells, and others. Case number 1 in Chapter 57 describes one condition in which there is a genetic deficiency of T cells due to mutation in the gene encoding adenosine deaminase. There are a variety of other conditions in which various components of the immune system are deficient due to mutations. Most of these are characterized by recurrent infections, which must be treated vigorously by, for example, administration of immunoglobulins (if these are deficient) and appropriate antibiotics.
This section considers only the plasma immunoglobulins, which are synthesized mainly in plasma cells. These are specialized cells of B-cell lineage that synthesize and secrete immunoglobulins into the plasma in response to exposure to a variety of antigens.
All Immunoglobulins Contain a Minimum of Two Light & Two Heavy Chains
Immunoglobulins contain a minimum of two identical light (L) chains (23 kDa) and two identical heavy (H) chains (53-75 kDa), held together as a tetramer (L2H2) by disulfide bonds. The structure of IgG is shown in Figure 50–14; it is Y-shaped, with binding of antigen occurring at both tips of the Y. Each chain can be divided conceptually into specific domains, or regions, that have structural and functional significance. The half of the light (L) chain toward the carboxyl terminal is referred to as the constant region (CL), while the amino terminal half is the variable region of the light chain (VL). Approximately one-quarter of the heavy (H) chain at the amino terminals is referred to as its variable region (VH), and the other three-quarters of the heavy chain are referred to as the constant regions (CH1, CH2, CH3) of that H chain. The portion of the immunoglobulin molecule that binds the specific antigen is formed by the amino terminal portions (variable regions) of both the H and L chains—that is, the VH and VL domains. The domains of the protein chains consist of two sheets of antiparallel distinct stretches of amino acids that bind antigen.
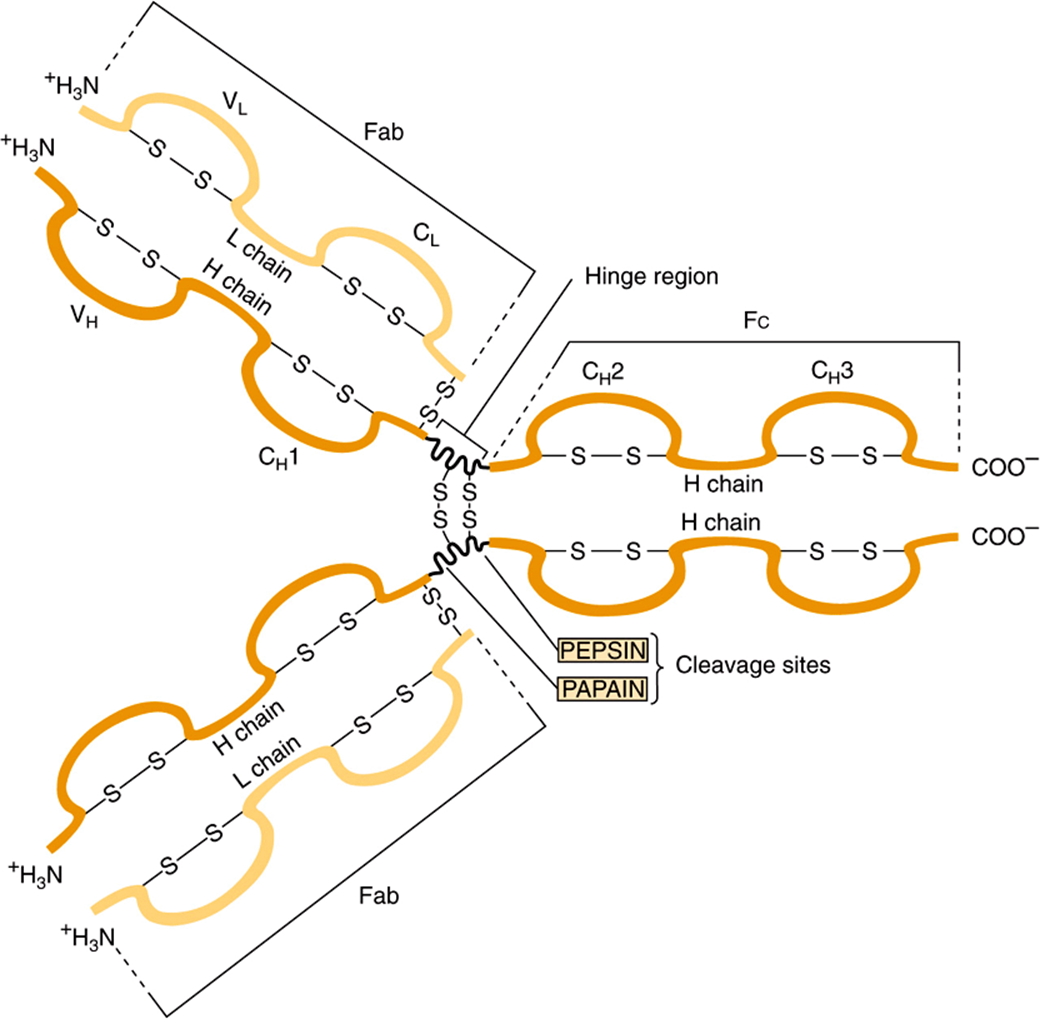
FIGURE 50–14 Structure of IgG. The molecule consists of two light (L) chains and two heavy (H) chains. Each light chain consists of a variable (VL) and a constant (CL) region. Each heavy chain consists of a variable region (VH) and a constant region that is divided into three domains (CH1, CH2, and CH3). The CH2 domain contains the complement-binding site and the CH3 domain contains a site that attaches to receptors on neutrophils and macrophages. The antigen-binding site is formed by the hypervariable regions of both the light and heavy chains, which are located in the variable regions of these chains (see Figure 50–10). The light and heavy chains are linked by disulfide bonds, and the heavy chains are also linked to each other by disulfide bonds. (Reproduced, with permission, from Parslow TG et al (editors): Medical Immunology, 10th ed. McGraw-Hill, 2001.)
As depicted in Figure 50–14 digestion of an immunoglobulin by the enzyme papain produces two antigen-binding fragments (Fab) and one crystallizable fragment (Fc), which is responsible for functions of immunoglobulins other than direct binding of antigens. Since there are two Fab regions, IgG molecules bind two molecules of antigen and are termed divalent. The site on the antigen to which an antibody binds is termed an antigenic determinant, or epitope. The area in which papain cleaves the immunoglobulin molecule—that is, the region between the CH1 and CH2 domains—is referred to as the “hinge region.” The hinge region confers flexibility and allows both Fab arms to move independently, thus helping them to bind to antigenic sites that may be variable distances apart (eg, on bacterial surfaces). Fc and hinge regions differ in the different classes of antibodies, but the overall model of antibody structure for each class is similar to that shown in Figure 50–14 for IgG.
All Light Chains Are Either Kappa or Lambda in Type
There are two general types of light chains, kappa (κ) and lambda (λ), which can be distinguished on the basis of structural differences in their CL regions. A given immunoglobulin molecule always contains two κ or two λ light chains—never a mixture of κ and λ. In humans, the κ chains are more frequent than λ chains in immunoglobulin molecules.
The Five Types of Heavy Chain Determine Immunoglobulin Class
Five classes of H chain have been found in humans (Table 50-9), distinguished by differences in their CH regions. They are designated λ, α, μ δ, and ε. The μ and ε chains each have four CH domains rather than the usual three. The type of H chain determines the class of immunoglobulin and thus its effector function. There are thus five immunoglobulin classes: IgG, IgA, IgM, IgD, and IgE. The biologic functions of these five classes are summarized in Table 50-10.
TABLE 50–9 Properties of Human Immunoglobulins
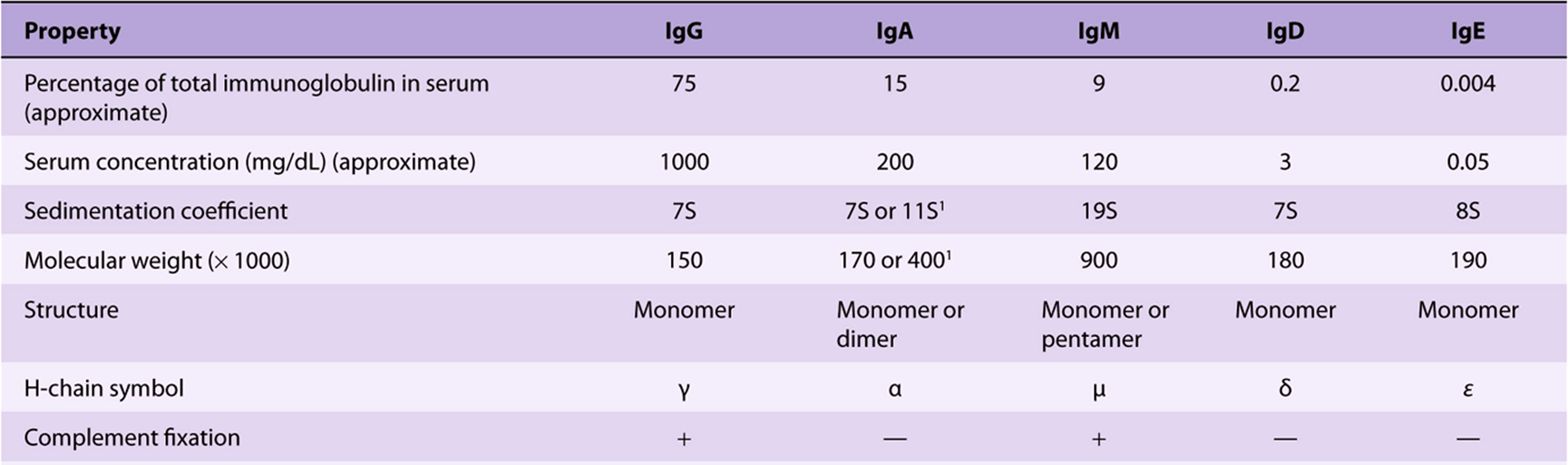
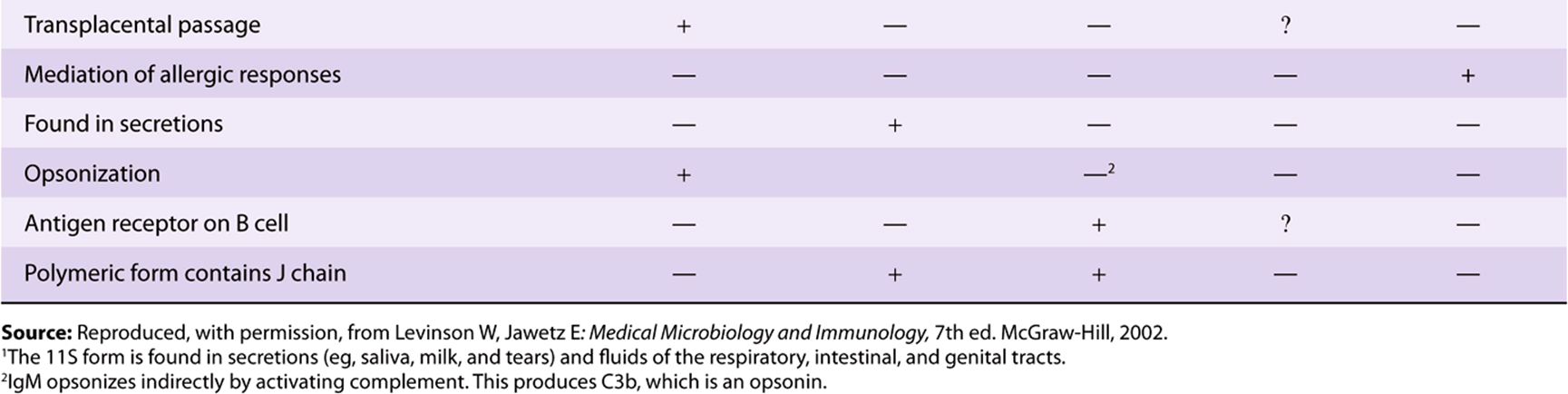
TABLE 50–10 Major Functions of Immunoglobulins
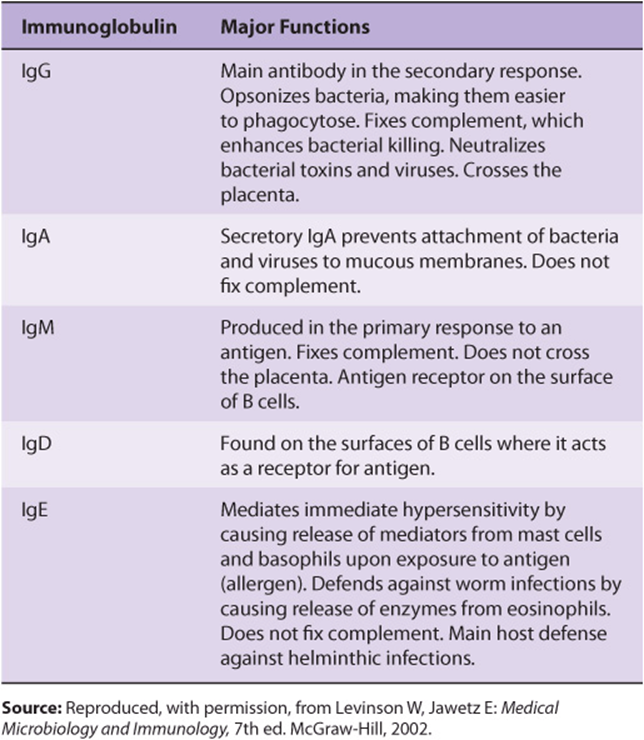
No Two Variable Regions Are Identical
The variable regions of immunoglobulin molecules consist of the VL and VH domains and are quite heterogeneous. In fact, no two variable regions from different humans have been found to have identical amino acid sequences. However, amino acid analyses have shown that the variable regions comprise relatively invariable regions and other hypervariable regions (Figure 50–15). L chains have three hypervariable regions (in VL) and H chains have four (in VH). These hypervariable regions comprise the antigen-binding site (located at the tips of the Y shown in Figure 50–14) and dictate the amazing specificity of antibodies. For this reason, hypervariable regions are also termed complementarity-determining regions (CDRs). About 5 to 10 amino acids in each hypervariable region (CDR) contribute to the antigen-binding site. CDRs are located on small loops of the variable domains, the surrounding polypeptide regions between the hypervariable regions being termed framework regions. CDRs from both VH and V, domains, brought together by folding of the polypeptide chains in which they are contained, form a single hypervariable surface comprising the antigen-binding site. Various combinations of H and L chain CDRs can give rise to many antibodies of different specificities, a feature that contributes to the tremendous diversity of antibody molecules and is termed combinatorial diversity. Large antigens interact with all of the CDRs of an antibody, whereas small ligands may interact with only one or a few CDRs that form a pocket or groove in the antibody molecule. The essence of antigen-antibody interactions is mutual complementarity between the surfaces of CDRs and epitopes. The interactions between antibodies and antigens involve noncovalent forces and bonds(electrostatic and van der Waals forces and hydrogen and hydrophobic bonds).
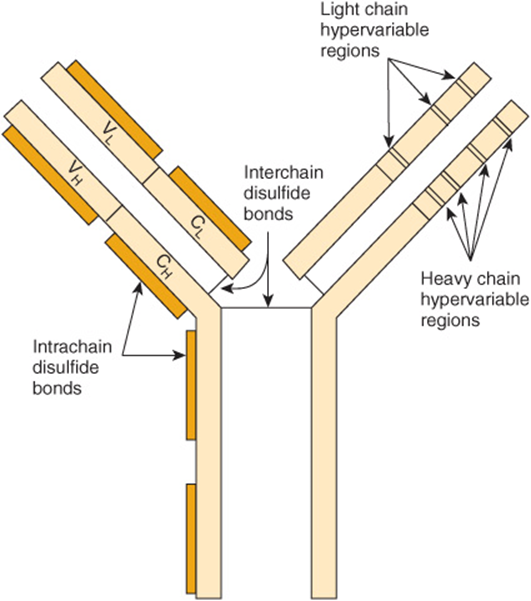
FIGURE 50–15 Schematic model of an IgG molecule showing approximate positions of the hypervariable regions in heavy and light chains. The antigen-binding site is formed by these hypervariable regions. The hypervariable regions are also called complementarity-determining regions (CDRs). (Modified and reproduced, with permission, from Parslow TG et al (editors): Medical Immunology, 10th ed. McGraw-Hill, 2001.)
The Constant Regions Determine Class-Specific Effector Functions
The constant regions of the immunoglobulin molecules, particularly the CH2 and CH3 (and CH4 of IgM and IgE), which constitute the Fc fragment, are responsible for the class-specific effector functions of the different immunoglobulin molecules (Table 50-9, bottom part), eg, complement fixation or transplacental passage.
Some immunoglobulins such as immune IgG exist only in the basic tetrameric structure, while others such as IgA and IgM can exist as higher order polymers of two, three (IgA), or five (IgM) tetrameric units (Figure 50–16).
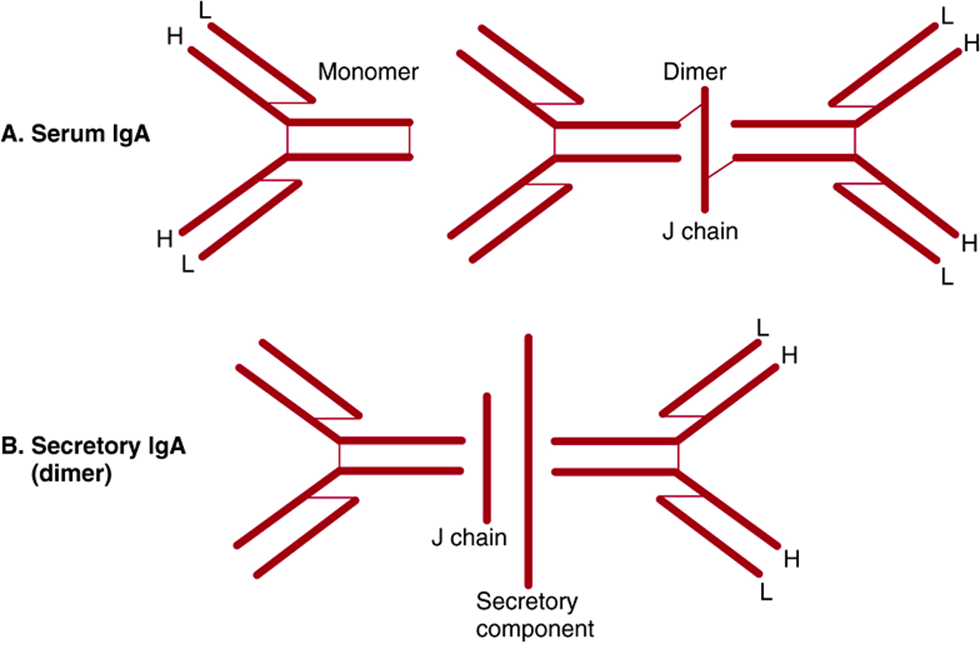
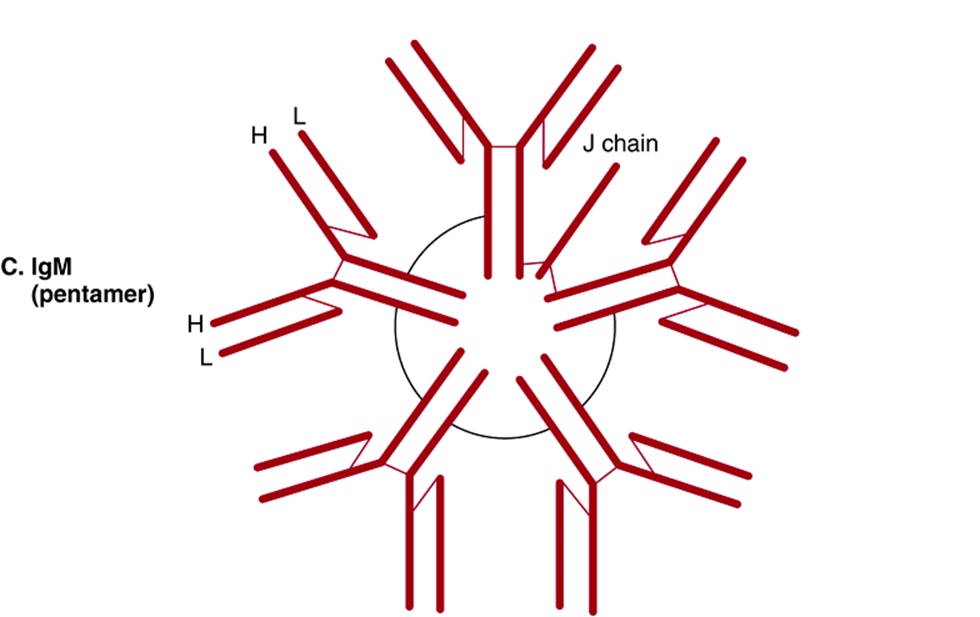
FIGURE 50–16 Schematic representation of serum IgA, secretory IgA, and IgM. Both IgA and IgM have a J chain, but only secretory IgA has a secretory component. Polypeptide chains are represented by thick lines; disulfide bonds linking different polypeptide chains are represented by thin lines. (Reproduced, with permission, from Parslow TG et al (editors): Medical Immunology, 10th ed. McGraw-Hill, 2001.)
The L chains and H chains are synthesized as separate molecules and are subsequently assembled within the B cell or plasma cell into mature immunoglobulin molecules, all of which are glycoproteins.
Both Light & Heavy Chains Are Products of Multiple Genes
Each immunoglobulin light chain is the product of at least three separate structural genes: a variable region (VL) gene, a joining region (J) gene (bearing no relationship to the J chain of IgA or IgM), and a constant region (CL)gene. Each heavy chain is the product of at least four different genes: a variable region (VH) gene, a diversity region (D) gene, a joining region (J) gene, and a constant region (CH) gene. Thus, the “one gene, one protein” concept is not valid. The molecular mechanisms responsible for the generation of the single immunoglobulin chains from multiple structural genes are discussed in Chapters 35 and 38.
Antibody Diversity Depends on Gene Rearrangements
Each person is capable of generating antibodies directed against perhaps 1 million different antigens. The generation of such immense antibody diversity depends upon a number of factors including the existence of multiple gene segments (V, C, J, and D segments), their recombinations (see Chapters 35 and 38), the combinations of different L and H chains, a high frequency of somatic mutations in immunoglobulin genes, and junctional diversity. The latter reflects the addition or deletion of a random number of nucleotides when certain gene segments are joined together and introduces an additional degree of diversity. Thus, the above factors ensure that a vast number of antibodiescan be synthesized from several hundred gene segments.
Class (Isotype) Switching Occurs During Immune Responses
In most humoral immune responses, antibodies with identical specificity but of different classes are generated in a specific chronologic order in response to the immunogen (immunizing antigen). For instance, antibodies of the IgM class normally precede molecules of the IgG class. The switch from one class to another is designated “class or isotype switching,” and its molecular basis has been investigated extensively. A single type of immunoglobulin light chain can combine with an antigen-specific μ chain to generate a specific IgM molecule. Subsequently, the same antigen-specific light chain combines with a γ chain with an identical VH region to generate an IgG molecule with antigen specificity identical to that of the original IgM molecule. The same light chain can also combine with an α heavy chain, again containing the identical VH region, to form an IgA molecule with identical antigen specificity. These three classes (IgM, IgG, and IgA) of immunoglobulin molecules against the same antigen have identical variable domains of both their light (VL) chains and heavy (VH) chains and are said to share an idiotype. (Idiotypes are the antigenic determinants formed by the specific amino acids in the hypervariable regions.) The different classes of these three immunoglobulins (called isotypes) are thus determined by their different CH regions, which are combined with the same antigen-specific VH regions.
Both Over-& Underproduction of Immunoglobulins May Result in Disease States
Disorders of immunoglobulins include increased production of specific classes of immunoglobulins or even specific immunoglobulin molecules, the latter by clonal tumors of plasma cells called myelomas. Multiple myeloma is a neoplastic condition; electrophoresis of serum or urine will usually reveal a large increase of one particular immunoglobulin or one particular light chain (the latter termed a Bence-Jones protein). Decreased production may be restricted to a single class of immunoglobulin molecules (eg, IgA or IgG) or may involve underproduction of all classes of immunoglobulins (IgA, IgD, IgE, IgG, and IgM). A severe reduction in synthesis of an immunoglobulin class due to a genetic abnormality can result in a serious immunodeficiency disease—for example, agammaglobulinemia, in which production of IgG is markedly affected—because of impairment of the body’s defense against microorganisms.
Hybridomas Provide Long-Term Sources of Highly Useful Monoclonal Antibodies
When an antigen is injected into an animal, the resulting antibodies are polyclonal, being synthesized by a mixture of B cells. Polyclonal antibodies are directed against a number of different sites (epitopes or determinants) on the antigen and thus are not monospecific. However, by means of a method developed by Kohler and Milstein, almost limitless amounts of a single monoclonal antibody specific for one epitope can be obtained.
The method involves cell fusion, and the resulting permanent cell line is called a hybridoma. Typically, B cells are obtained from the spleen of a mouse (or other suitable animal) previously injected with an antigen or mixture of antigens (eg, foreign cells). The B cells are mixed with mouse myeloma cells and exposed to polyethylene glycol, which causes cell fusion. A summary of the principles involved in generating hybridoma cells is given in Figure 50–17. Under the conditions used, only the hybridoma cells multiply in cell culture. This involves plating the hybrid cells into hypoxanthine-aminopterin-thymidine (HAT)-containing medium at a concentration such that each dish contains approximately one cell. Thus, a clone of hybridoma cells multiplies in each dish. The culture medium is harvested and screened for antibodies that react with the original antigen or antigens. If the immunogen is a mixture of many antigens (eg, a cell membrane preparation), an individual culture dish will contain a clone of hybridoma cells synthesizing a monoclonal antibody to one specific antigenic determinant of the mixture. By harvesting the media from many culture dishes, a battery of monoclonal antibodies can be obtained, many of which are specific for individual components of the immunogenic mixture. The hybridoma cells can be frozen and stored and subsequently thawed when more of the antibody is required; this ensures its long-term supply. The hybridoma cells can also be grown in the abdomen of mice, providing relatively large supplies of antibodies. Attempts to produce human monoclonal antibodies are underway.
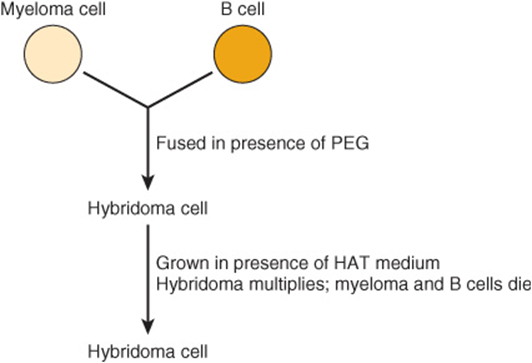
FIGURE 50–17 Scheme of production of a hybridoma cell. The myeloma cells are immortalized, do not produce antibody, and are HGPRT– (rendering the salvage pathway of purine synthesis [see Chapter 33] inactive). The B cells are not immortalized, each produces a specific antibody, and they are HGPRT+. Polyethylene glycol (PEG) stimulates cell fusion. The resulting hybridoma cells are immortalized (via the parental myeloma cells), produce antibody, and are HGPRT+ (both latter properties gained from the parental B cells). The B cells will die in the medium because they are not immortalized. In the presence of HAT, the myeloma cells will also die since the aminopterin in HAT suppresses purine synthesis by the de novo pathway by inhibiting reutilization of tetrahydrofolate (see Chapter 33). However, the hybridoma cells will survive, grow (because they are HGPRT+), and—if cloned—produce monoclonal antibody. (HAT, hypoxanthine, aminopterin, and thymidine; HGPRT, hypoxanthineguanine phosphoribosyl transferase.)
Because of their specificity, monoclonal antibodies have become extremely useful reagents in many areas of biology and medicine. For example, they can be used to measure the amounts of many individual proteins (eg, plasma proteins), to determine the nature of infectious agents (eg, types of bacteria), and to subclassify both normal (eg, lymphocytes) and tumor cells (eg, leukemic cells). In addition, they are being used to direct therapeutic agents to tumor cells and also to accelerate removal of drugs from the circulation when they reach toxic levels (eg, digoxin).
For therapeutic use in humans, monoclonal antibodies made in mice can be humanized. This can be achieved by attaching the CDRs (the sites that bind antigens) onto appropriate sites in a human immunoglobulin molecule. This produces an antibody that is very similar to a human antibody, thus markedly lessening immunogenicity and the chances of an anaphylactic reaction.
The Complement System Comprises Some 20 Plasma Proteins & Is Involved in Cell Lysis, Inflammation, & Other Processes
Plasma contains approximately 20 proteins that are members of the complement system. This system was discovered when it was observed that the addition of fresh serum containing antibodies directed to a bacterium caused its lysis. Unlike antibodies, the factor was labile when heated at 56°C. Subsequent work has resolved the proteins of the system and how they function; most have been cloned and sequenced. The complement system is involved in the ability to lyse various cells, but also in aspects of inflammation (eg, chemotaxis and phagocytosis) and in the clearance of antigen-antibody complexes from the circulation. Deficiencies of various components of the system due to mutations cause complement deficiency disorders. The details of this system are relatively complex, and a textbook of immunology should be consulted. The basic concept is that the normally inactive proteins of the system, when triggered by various stimuli, become activated by proteolysis and interact in a specific sequence with one or more of the other proteins of the system. The overall result of activation of the pathway is cell lysis and generation of peptide or polypeptide fragments that are involved in aspects of inflammation. The complement system resembles blood coagulation (Chapter 51) in that it involves both conversion of inactive precursors to active products by proteases and a cascade with amplification.
SUMMARY
![]() Plasma contains many proteins with a variety of functions. Most are synthesized in the liver and are glycosylated.
Plasma contains many proteins with a variety of functions. Most are synthesized in the liver and are glycosylated.
![]() Albumin, which is not glycosylated, is the major protein and is the principal determinant of intravascular osmotic pressure; it also binds many ligands, such as drugs and bilirubin.
Albumin, which is not glycosylated, is the major protein and is the principal determinant of intravascular osmotic pressure; it also binds many ligands, such as drugs and bilirubin.
![]() Haptoglobin binds extracorpuscular hemoglobin, prevents its loss into the kidney and urine, and hence preserves its iron for reutilization.
Haptoglobin binds extracorpuscular hemoglobin, prevents its loss into the kidney and urine, and hence preserves its iron for reutilization.
![]() Transferrin binds iron, transporting it to sites where it is required. Ferritin provides an intracellular store of iron. The regulation of body iron levels involves a battery of proteins, some of which—such as ferroportin and hepcidin—have only been discovered relatively recently. Iron deficiency anemia is a very prevalent disorder. Hereditary hemochromatosis is a genetic disease involving excess absorption of iron; it is discussed in Chapter 57 (Case 10). A number of laboratory tests are available for assessing the status of iron (eg, excess or deficiency) in the human body, and many different proteins are involved in different aspects of its metabolism.
Transferrin binds iron, transporting it to sites where it is required. Ferritin provides an intracellular store of iron. The regulation of body iron levels involves a battery of proteins, some of which—such as ferroportin and hepcidin—have only been discovered relatively recently. Iron deficiency anemia is a very prevalent disorder. Hereditary hemochromatosis is a genetic disease involving excess absorption of iron; it is discussed in Chapter 57 (Case 10). A number of laboratory tests are available for assessing the status of iron (eg, excess or deficiency) in the human body, and many different proteins are involved in different aspects of its metabolism.
![]() Ceruloplasmin contains substantial amounts of copper, but albumin appears to be more important with regard to its transport. Both Wilson disease and Menkes disease, which reflect abnormalities of copper metabolism, have been found to be due to mutations in genes encoding copper-binding P-type ATPases.
Ceruloplasmin contains substantial amounts of copper, but albumin appears to be more important with regard to its transport. Both Wilson disease and Menkes disease, which reflect abnormalities of copper metabolism, have been found to be due to mutations in genes encoding copper-binding P-type ATPases.
![]() α1-Antitrypsin is the major serine protease inhibitor of plasma, in particular, inhibiting the elastase of neutrophils. Genetic deficiency of this protein is a cause of emphysema and can also lead to liver disease.
α1-Antitrypsin is the major serine protease inhibitor of plasma, in particular, inhibiting the elastase of neutrophils. Genetic deficiency of this protein is a cause of emphysema and can also lead to liver disease.
![]() α2-Macroglobulin is a major plasma protein that neutralizes many proteases and targets certain cytokines to specific organs.
α2-Macroglobulin is a major plasma protein that neutralizes many proteases and targets certain cytokines to specific organs.
![]() Immunoglobulins play a key role in the defense mechanisms of the body, as do proteins of the complement system. Some of the principal features of these proteins are briefly described.
Immunoglobulins play a key role in the defense mechanisms of the body, as do proteins of the complement system. Some of the principal features of these proteins are briefly described.
REFERENCES
Andrew NC: Forging a field: the golden age of iron biology. Blood 2008;112:219.
Burtis CA, Ashwood EA, Bruns DE (editors): Tietz Textbook of Clinical Chemistry and Molecular Diagnostics, 4th ed. Elsevier Saunders, 2006. (Chapters 20, 26, and 31 give extensive coverage of plasma proteins, complement proteins, immunoglobulins, C-reactive protein, hemoglobin, iron, and bilirubin).
Craig WY, Ledue TB, Ritchie RF: Plasma Proteins: Clinical Utility and Interpretation. Foundation for Blood Research, 2008.
Fauci AS, Branwald E, Kasper DL, et al: Harrison’s Principles of Internal Medicine, 17th ed. McGraw-Hill, 2008 (Chapters 58, 98, and 308 contain coverage of anemia and polycythemia, iron deficiency and other hypoproliferative anemias, and an introduction to the immune system).
Ganz T: Iron homeostasis: fitting the puzzle pieces together. Cell Metab 2008;7:288.
Hentz MW, Muckenthaler MU, Gali B, et al: Two to tango: regulation of mammalian iron metabolism. Cell 2010;142:24.
Lab Tests Online: http://www.labtestsonline.org/ (A comprehensive web site provided by the American Association of Clinical Chemists that provides information on the measurement and significance of the various plasma proteins discussed in this Chapter, and also on most other lab tests.)
Levinson W: Review of Medical Microbiology and Immunology, 11th ed. Appleton & Lange, 2010. (Good description of the basics of Immunology).
Murphy KM, Travers P, Walport M: Janeway’s Immunobiology, 7th ed. Garland Science Publishing, 2007.
Schaller H, Gerber S, Kaempfer U, et al: Human Blood Plasma Proteins: Structure and Function. Wiley, 2008.