Harper’s Illustrated Biochemistry, 29th Edition (2012)
SECTION VI. Special Topics
Chapter 51. Hemostasis & Thrombosis
Peter L. Gross, MD, MSc, FRCP(C), Robert K. Murray, MD, PhD, & Margaret L. Rand, PhD
OBJECTIVES
After studying this chapter, you should be able to:
![]() Understand the significance of hemostasis and thrombosis in health and disease.
Understand the significance of hemostasis and thrombosis in health and disease.
![]() Outline the pathways of coagulation that result in the formation of fibrin.
Outline the pathways of coagulation that result in the formation of fibrin.
![]() Identify the vitamin K-dependent coagulation factors.
Identify the vitamin K-dependent coagulation factors.
![]() Provide examples of genetic disorders that lead to bleeding.
Provide examples of genetic disorders that lead to bleeding.
![]() Describe the process of fibrinolysis.
Describe the process of fibrinolysis.
![]() Outline the steps leading to platelet aggregation.
Outline the steps leading to platelet aggregation.
![]() Identify the antiplatelet drugs and their mode of inhibition of platelet aggregation.
Identify the antiplatelet drugs and their mode of inhibition of platelet aggregation.
BIOMEDICAL IMPORTANCE
Basic aspects of the proteins of the blood coagulation system and of fibrinolysis are described in this chapter. Some fundamental aspects of platelet biology are also presented. Hemorrhagic and thrombotic states can cause serious medical emergencies, and thromboses in the coronary and cerebral arteries are major causes of death in many parts of the world. Rational management of these conditions requires a clear understanding of the bases of blood coagulation, fibrinolysis, and platelet activation.
HEMOSTASIS&THROMBOSIS HAVE THREE COMMON PHASES
Hemostasis is the cessation of bleeding from a cut or severed vessel, whereas thrombosis occurs when the endothelium lining blood vessels is damaged or removed (eg, upon rupture of an atherosclerotic plaque). These processes involve blood vessels, platelet aggregation, and plasma proteins that cause formation or dissolution of platelet aggregates and fibrin.
In hemostasis, there is initial vasoconstriction of the injured vessel, causing diminished blood flow distal to the injury. Then, hemostasis and thrombosis share three phases:
1. Formation of a loose and temporary platelet aggregate at the site of injury. Platelets bind to collagen at the site of vessel wall injury, and form thromboxane A2 and release ADP, which activate other platelets flowing by the vicinity of the injury. (The mechanism of platelet activation is described below.) Thrombin, formed during coagulation at the same site, causes further platelet activation. Upon activation, platelets change shape and, in the presence of fibrinogen and/or von Willebrand factor, aggregate to form the hemostatic plug (in hemostasis) or thrombus (in thrombosis).
2. Formation of a fibrin mesh that binds to the platelet aggregate, forming a more stable hemostatic plug or thrombus.
3. Partial or complete dissolution of the hemostatic plug or thrombus by plasmin.
There Are Three Types of Thrombi
Three types of thrombi or clots are distinguished. All three contain fibrin in various proportions.
1. The white thrombus is composed of platelets and fibrin and is relatively poor in erythrocytes. It forms at the site of an injury or abnormal vessel wall, particularly in areas where blood flow is rapid (arteries).
2. The red thrombus consists primarily of red cells and fibrin. It morphologically resembles the clot formed in a test tube and may form in vivo in areas of retarded blood flow or stasis (eg, veins) with or without vascular injury, or it may form at a site of injury or in an abnormal vessel in conjunction with an initiating platelet plug.
3. A third type is fibrin deposits in very small blood vessels or capillaries.
We shall first describe the coagulation pathway leading to the formation of fibrin. Then, we shall briefly describe some aspects of the involvement of platelets and blood vessel walls in the overall process. This separation of clotting factors and platelets is artificial since both play intimate and often mutually interdependent roles in hemostasis and thrombosis, but it facilitates description of the overall processes involved.
Both Extrinsic & Intrinsic Pathways Result in the Formation of Fibrin
Two pathways lead to fibrin clot formation: the extrinsic and the intrinsic pathways. These pathways are not independent, as previously thought. However, this artificial distinction is retained in the following text to facilitate their description.
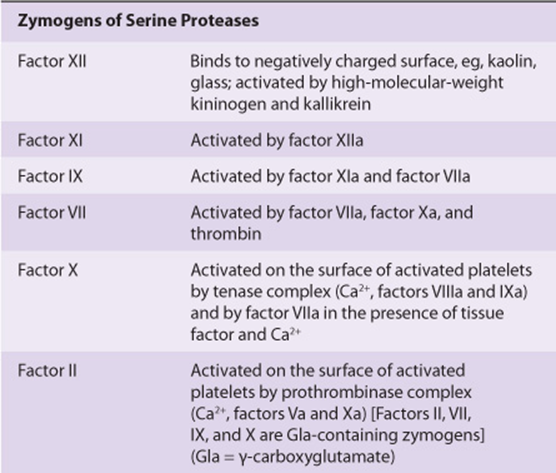
Initiation of fibrin clot formation in response to tissue injury is carried out by the extrinsic pathway. The intrinsic pathway is activated by negatively charged surfaces in vitro, eg, glass. Both pathways lead to activation of prothrombin to thrombin and the thrombin-catalyzed cleavage of fibrinogen to form the fibrin clot. The pathways are complex and involve many different proteins (Figures 51-1 and 51-2; Table 51-1). In general, as shown in Table 51-2, these proteins can be classified into five types: (1) zymogens of serine-dependent proteases, which become activated during the process of coagulation; (2) cofactors; (3) fibrinogen; (4) a transglutaminase, which stabilizes the fibrin clot; and (5) regulatory and other proteins.
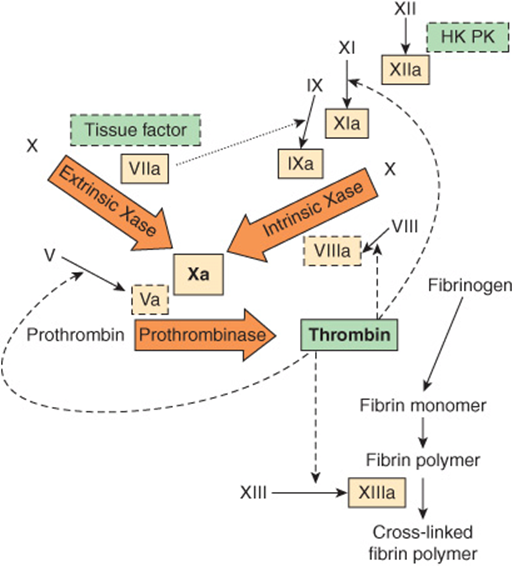
FIGURE 51–1 The pathways of blood coagulation, with the extrinsic pathway indicated at the top left and the intrinsic pathway at the top right. The pathways converge in the formation of factor Xa and culminate in the formation of cross-linked fibrin. Complexes of tissue factor and factor VIIa activate not only factor X (extrinsic Xase [tenase]) but also factor IX in the intrinsic pathway (dotted arrow). In addition, thrombin feedback activates at the sites indicated (dashed arrows) and also activates factor VII to factor VIIa (not shown). The three predominant complexes, extrinsic Xase, intrinsic Xase, and prothrombinase, are indicated in the arrows; the reactions require anionic procoagulant phospholipid membrane and calcium. Activated proteases are in solid-outlined boxes; active cofactors are in dash-outlined boxes and inactive factors are not in boxes. (HK, high-molecular-weight kininogen; PK, prekallikrein.)
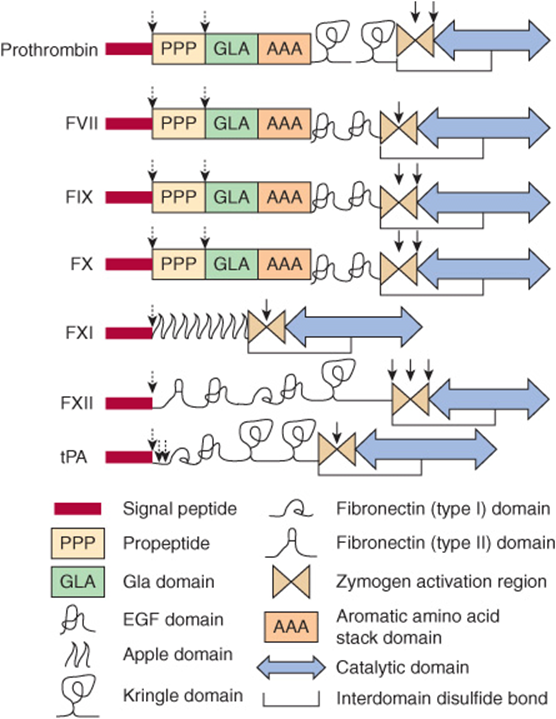
FIGURE 51–2 The structural domains of selected proteins involved in coagulation and fibrinolysis. The domains are as identified at the bottom of the figure and include signal peptide, propeptide, Gla (γ-carboxyglutamate) domain, epidermal growth factor (EGF) domain, apple domain, kringle domain, fibronectin (types I and II) domain, the zymogen activation region, aromatic amino acid stack, and the catalytic domain. Interdomain disulfide bonds are indicated, but numerous intradomain disulfide bonds are not. Sites of proteolytic cleavage in synthesis or activation are indicated by arrows (dashed and solid, respectively). FVII, factor VII; FIX, factor IX; FX, factor X, FXI; factor XI; FXII, factor XII; tPA, tissue plasminogen activator. (Adapted, with permission, from Furie B, Furie BC: The molecular basis of blood coagulation. Cell 1988;53:505.)
TABLE 51–1 Numerical System for Nomenclature of Blood Clotting Factors
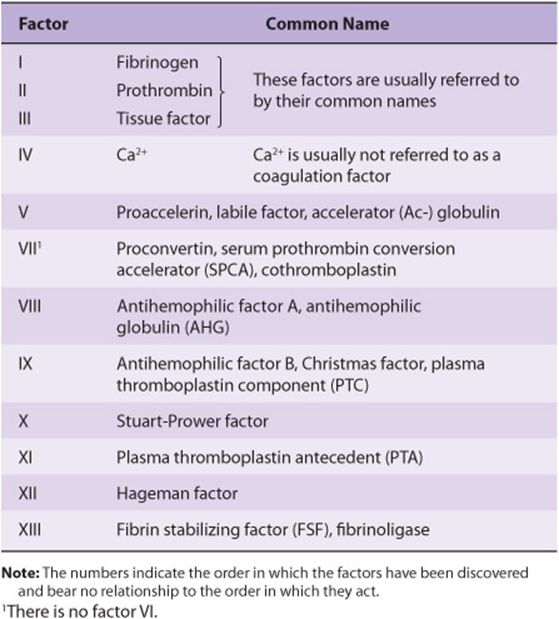
TABLE 51–2 The Functions of the Proteins Involved in Blood Coagulation

The Extrinsic Pathway Leads to Activation of Factor X
The extrinsic pathway involves tissue factor, factors VII and X, and Ca2+ and results in the production of factor Xa (by convention, activated clotting factors are referred to by use of the suffix a). It is initiated at the site of tissue injury with the exposure of tissue factor (Figure 51–1), located in the subendothelium and on activated monocytes. Tissue factor interacts with and activates factor VII (53 kDa, a zymogen containing vitamin K-dependent γ -carboxyglutamate [Gla] residues; see Chapter 44), synthesized in the liver. It should be noted that in the Gla-containing zymogens (factors II, VII, IX, and X), the Gla residues in the amino terminal regions of the molecules serve as high-affinity binding sites for Ca2+. Tissue factor acts as a cofactor for factor VIIa, enhancing its enzymatic activity to activate factor X (56 kDa). The reaction by which factor X is activated requires the assembly of components, termed the extrinsic tenase complex, on a cell membrane surface exposing the procoagulant phospholipid phosphatidylserine; these components are Ca2+, tissue factor, factor VIIa, and factor X. Factor VIIa cleaves an Arg-Ile bond in factor X to produce the two-chain serine protease, factor Xa. Tissue factor and factor VIIa also activate factor IX in the intrinsic pathway. Indeed, the formation of complexes between tissue factor and factor VIIa is now considered to be the key process involved in initiation of blood coagulation in vivo.
Tissue factor pathway inhibitor (TFPI) is a major physiologic inhibitor of coagulation. It is a protein that circulates in the blood associated with lipoproteins. TFPI directly inhibits factor Xa by binding to the enzyme near its active site. This factor Xa-TFPI complex then inhibits the factor VIIa-tissue factor complex.
The Intrinsic Pathway Also Leads to Activation of Factor X
The activation of factor Xa is the major site where the intrinsic and extrinsic pathways converge (Figure 51–1). The intrinsic pathway (Figure 51–1) involves factors XII, XI, IX, VIII, and X as well as prekallikrein, high-molecular-weight (HMW) kininogen, Ca2+, and phospholipid. It results in the production of factor Xa that is cleaved by the tenase complex, with factor IXa as the serine protease and factor VIIIa as the cofactor, of the intrinsic pathway. Activation of factor X provides an important link between the intrinsic and extrinsic pathways.
The intrinsic pathway can be initiated with the “contact phase” in which prekallikrein, HMW kininogen, factor XII, and factor XI are exposed to a negatively charged activating surface. Kaolin can be used for in vitro tests as an initiator of the intrinsic pathway. When the components of the contact phase assemble on the activating surface, factor XII is activated to factor XIIa upon proteolysis by kallikrein. This factor XIIa, generated by kallikrein, attacks prekallikrein to generate more kallikrein, setting up a reciprocal activation. Factor XIIa, once formed, activates factor XI to XIa and also releases bra-dykinin (a nonapeptide with potent vasodilator action) from HMW kininogen.
Factor XIa in the presence of Ca2+ activates factor IX (55 kDa, a Gla-containing zymogen), to the serine protease, factor IXa. This, in turn, also cleaves an Arg-Ile bond in factor X to produce factor Xa. This latter reaction requires the assembly of components, called the intrinsic tenase complex, on a membrane surface: Ca2+ and factor VIIIa, as well as factors IXa and X.
Factor VIII (330 kDa), a circulating glycoprotein, is not a protease precursor but a cofactor that serves as a receptor for factors IXa and X on the platelet surface. Factor VIII is activated by minute quantities of thrombin to form factor VIIIa, which is in turn inactivated upon further cleavage by thrombin.
The role of the initial steps of the intrinsic pathway in initiating coagulation has been called into question because patients with a hereditary deficiency of factor XII, prekallikrein or HMW kininogen do not exhibit bleeding problems. Similarly, patients with a deficiency of factor XI may not have bleeding problems. The intrinsic pathway largely serves to amplify factor Xa and ultimately thrombin formation, through feedback mechanisms (see below). The intrinsic pathway may also be important in fibrinolysis (see below) since kallikrein, factor XIIa, and factor XIa can cleave plasminogen and kallikrein can activate single-chain urokinase.
Factor Xa Leads to Activation of Prothrombin to Thrombin
Factor Xa, produced by either the extrinsic or the intrinsic pathway, activates prothrombin (factor II) to thrombin (factor IIa) (Figure 51–1).
The activation of prothrombin, like that of factor X, occurs on a membrane surface and requires the assembly of a prothrombinase complex, consisting of Ca2+, factor Va, factor Xa, and prothrombin. The assembly of the prothrombinase and tenase complexes takes place on the membrane surface of platelets activated to expose the acidic (anionic) phospholipid phosphatidylserine, which is normally on the internal side of the plasma membrane of resting, nonactivated platelets.
Factor V (330 kDa), a glycoprotein with homology to factor VIII and ceruloplasmin, is synthesized in the liver, spleen, and kidney and is found in platelets as well as in plasma. It functions as a cofactor in a manner similar to that of factor VIII in the tenase complex. When activated to factor Va by traces of thrombin, it binds specifically to the platelet membrane (Figure 51–3) and forms a complex with factor Xa and prothrombin. It is subsequently inactivated by activated protein C (see below), thereby providing a means of limiting the activation of prothrombin to thrombin. Prothrombin (72 kDa; Figure 51–3) is a single-chain glycoprotein synthesized in the liver. The amino terminal region of prothrombin (Figure 51–2) contains 10 Gla residues, and the serine-dependent active protease site is in the catalytic domain close to the carboxyl terminal region of the molecule. Upon binding to the complex of factors Va and Xa on the platelet membrane (Figure 51–3), prothrombin is cleaved by factor Xa at two sites to generate the active, two-chain thrombin molecule, which is then released from the platelet surface.
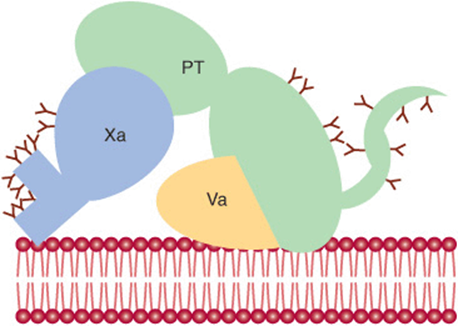
FIGURE 51–3 Diagrammatic representation (not to scale) of the binding of factors Va, Xa, and prothrombin (PT) to the plasma membrane of the activated platelet. A central theme in blood coagulation is the assembly of protein complexes on membrane surfaces. Gamma-carboxyglutamate residues (indicated by Y) on vitamin K-dependent proteins bind calcium and contribute to the exposure of membrane binding sites on these proteins. (Adapted, with permission, from Furie B, Furie BC: The molecular basis of blood coagulation. Cell 1988;53:505.)
Conversion of Fibrinogen to Fibrin Is Catalyzed by Thrombin
Thrombin, produced by the prothrombinase complex, in addition to having a potent stimulatory effect on platelets (see below), converts fibrinogen to fibrin (Figure 51–1). Fibrinogen (factor I, 340 kDa; see Figures 51-1 and 51-4; Tables 51-1 and 51-2) is a soluble plasma glycoprotein that consists of three nonidentical pairs of polypeptide chains (Aα, Bβ,γ)2, covalently linked by disulfide bonds. The Bβ and γ chains contain asparagine-linked complex oligosaccharides. All three chains are synthesized in the liver; the three genes are on the same chromosome, and their expression is coordinately regulated in humans. The amino terminal regions of the six chains are held in close proximity by a number of disulfide bonds, while the carboxyl terminal regions are spread apart, giving rise to a highly asymmetric, elongated molecule (Figure 51–4). The Aα and Bβ portions of the A and B chains, designated fibrinopeptide A (FPA) and fibrinopeptide B (FPB), respectively, at the amino terminal ends of the chains, bear excess negative charges as a result of the presence of aspartate and glutamate residues, as well as an unusual tyrosine O-sulfate in FPB. These negative charges contribute to the solubility of fibrinogen in plasma and also serve to prevent aggregation by causing electrostatic repulsion between fibrinogen molecules.
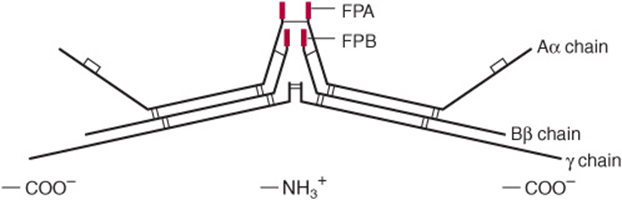
FIGURE 51–4 Diagrammatic representation (not to scale) of fibrinogen showing pairs of Aα, Bβ, and γ chains linked by disulfide bonds. (FPA, fibrinopeptide A; FPB, fibrinopeptide B.)
Thrombin (34 kDa), a serine protease formed by the prothrombinase complex, hydrolyzes the four Arg-Gly bonds between the fibrinopeptides and the α and β portions of the Aα and Bβ chains of fibrinogen (Figure 51–5A). The release of the fibrinopeptides by thrombin generates fibrin monomer, which has the subunit structure (α, β, γ)2. Since FPA and FPB contain only 16 and 14 residues, respectively, the fibrin molecule retains 98% of the residues present in fibrinogen. The removal of the fibrinopeptides exposes binding sites that allow the molecules of fibrin monomers to aggregate spontaneously in a regularly staggered array, forming an insoluble fibrin clot. This initial fibrin clot is rather weak, held together only by the noncovalent association of fibrin monomers.
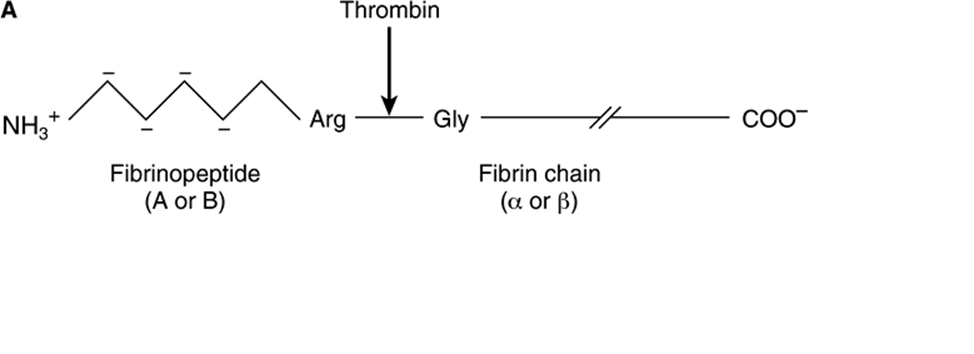
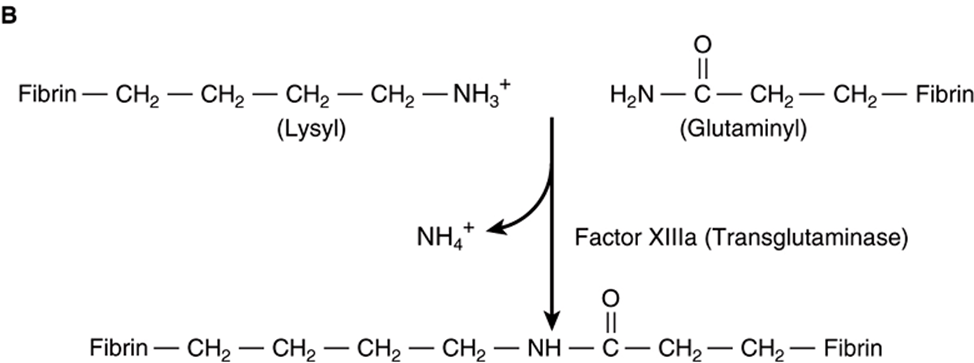
FIGURE 51–5 Formation of a fibrin clot. (A) Thrombin-induced cleavage of Arg-Gly bonds of the Aα and Bβ chains of fibrinogen to produce fibrinopeptides (left-hand side) and the α and β chains of fibrin monomer (right-hand side). (B) Cross-linking of fibrin molecules by activated factor XIII (factor XIIIa).
In addition to converting fibrinogen to fibrin, thrombin also converts factor XIII to factor XIIIa. The latter is a highly specific transglutaminase that covalently cross-links fibrin molecules by forming peptide bonds between the amide groups of glutamine and the ε-amino groups of lysine residues (Figure 51–5B), yielding a more stable fibrin clot with increased resistance to proteolysis. This fibrin mesh serves to stabilize the hemostatic plug or thrombus.
Levels of Circulating Thrombin Are Carefully Controlled
Once active thrombin is formed in the course of hemostasis or thrombosis, its concentration must be carefully controlled to prevent further fibrin formation or platelet activation. This is achieved in two ways. Thrombin circulates as its inactive precursor, prothrombin, which is activated as a result of a cascade of enzymatic reactions, each converting an inactive zymogen to an active enzyme and leading finally to the conversion of prothrombin to thrombin (Figure 51–1). At each point in the cascade, feedback mechanisms produce a delicate balance of activation and inhibition. The concentration of factor XII in plasma is approximately 30 μg/mL, while that of fibrinogen is 3 mg/mL, with intermediate clotting factors increasing in concentration as one proceeds down the cascade, showing that the clotting cascade provides amplification. The second means of controlling thrombin activity is the inactivation of any thrombin formed by circulating inhibitors, the most important of which is antithrombin (see below).
The Activity of Antithrombin, an Inhibitor of Thrombin, Is Increased by Heparin
Four naturally occurring thrombin inhibitors exist in normal plasma. The most important is antithrombin, which contributes approximately 75% of the antithrombin activity. Antithrombin can also inhibit the activities of factors IXa, Xa, XIa, XIIa, and VIIa complexed with tissue factor. α2-Macroglobulin contributes most of the remainder of the antithrombin activity, with heparin cofactor II and α1-antitrypsin acting as minor inhibitors under physiologic conditions.
The endogenous activity of antithrombin is greatly potentiated by the presence of sulfated glycosaminoglycans (heparans) (Chapter 48). These bind to a specific cationic site of antithrombin, inducing a conformational change and promoting its binding to thrombin as well as to its other substrates. This is the basis for the use of heparin, a derivatized heparan, in clinical medicine to inhibit coagulation. The anticoagulant effects of heparin can be antagonized by strongly cationic polypeptides such as protamine, which bind strongly to heparin, thus inhibiting its binding to antithrombin.
Low-molecular-weight heparins (LMWHs), derived from enzymatic or chemical cleavage of unfractionated heparin, are finding increasing clinical use. They can be administered subcutaneously at home, have greater bioavailability than unfractionated heparin, and do not need frequent laboratory monitoring.
Individuals with inherited deficiencies of antithrombin are prone to develop venous thrombosis, providing evidence that antithrombin has a physiologic function and that the coagulation system in humans is normally in a dynamic state.
Thrombin is involved in an additional regulatory mechanism that operates in coagulation. It combines with thrombomodulin, a glycoprotein present on the surfaces of endothelial cells. The complex activates protein C on the endothelial protein C receptor. In combination with protein S, activated protein C (APC) degrades factors Va and VIIIa, limiting their actions in coagulation. A genetic deficiency of either protein C or protein S can cause venous thrombosis. Furthermore, patients with factor V Leiden (which has a glutamine residue in place of an arginine at position 506) have an increased risk of venous thrombotic disease because factor V Leiden is resistant to inactivation by APC. This condition is termed APC resistance.
Coumarin Anticoagulants Inhibit the Vitamin K-Dependent Carboxylation of Factors II, VII, IX, & X
The coumarin drugs (eg, warfarin), which are used as anticoagulants, inhibit the vitamin K-dependent carboxylation of Glu to Gla residues (see Chapter 44) in the amino terminal regions of factors II, VII, IX, and X and also proteins C and S. These proteins, all of which are synthesized in the liver, are dependent on the Ca2+-binding properties of the Gla residues for their normal function in the coagulation pathways. The coumarins act by inhibiting the reduction of the quinone derivatives of vitamin K to the active hydroquinone forms (Chapter 44). Thus, the administration of vitamin K will bypass the coumarin-induced inhibition and allow the post-translational modification of carboxylation to occur. Reversal of coumarin inhibition by vitamin K requires 12-24 h, whereas reversal of the anticoagulant effects of heparin by protamine is almost instantaneous.
Heparin and warfarin are widely used in the treatment of thrombotic and thromboembolic conditions, such as deep vein thrombosis and pulmonary embolism. Heparin is administered first, because of its prompt onset of action, whereas warfarin takes several days to reach full effect. Their effects are closely monitored by use of appropriate tests of coagulation (see below) because of the risk of producing hemorrhage.
New oral inhibitors of thrombin (dabigatran) or of factor Xa (rivaroxaban and others) are also used in the treatment of thrombotic conditions.
There Are Several Hereditary Bleeding Disorders, Including Hemophilia A
Inherited deficiencies of the clotting system that result in bleeding are found in humans. The most common is deficiency of factor VIII, causing hemophilia A, an X chromosome-linked disease. Hemophilia B, also X chromosome-linked, is due to a deficiency of factor IX and has recently been identified as the form of hemophilia that played a major role in the history of the royal families of Europe; its clinical features are almost identical to those of hemophilia A, but the conditions can be separated on the basis of specific assays that distinguish between the two factors.
The gene for human factor VIII has been cloned and is one of the largest so far studied, measuring 186 kb in length and containing 26 exons. A variety of mutations in the factor VIII and IX genes have been detected leading to diminished activities of the factor VIII and IX proteins; these include partial gene deletions and point and missense mutations. Prenatal diagnosis by DNA analysis after chorionic villus sampling is now possible.
In the past, treatment for patients with hemophilia A and B consisted of administration of cryoprecipitates (enriched in factor VIII) prepared from individual donors or lyophilized factor VIII or IX concentrates prepared from very large plasma pools. It is now possible to prepare factors VIII and IX by recombinant DNA technology. Such preparations are free of contaminating viruses (eg, hepatitis A, B, C, or HIV-1) found in human plasma, but are expensive; their use may increase if cost of production decreases.
The most common hereditary bleeding disorder is von Willebrand disease, with a prevalence of up to 1% of the population. It results from a deficiency or defect in von Willebrand factor, a large multimeric glycoprotein that is secreted by endothelial cells and platelets into the plasma, where it stabilizes factor VIII. von Willebrand factor also promotes platelet adhesion at sites of vessel wall injury (see below).
Fibrin Clots Are Dissolved by Plasmin
As stated above, the coagulation system is normally in a state of dynamic equilibrium in which fibrin clots are constantly being laid down and dissolved. This latter process is termed fibrinolysis. Plasmin, the serine protease mainly responsible for degrading fibrin and fibrinogen, circulates in the form of its inactive zymogen, plasminogen (90 kDa), and any small amounts of plasmin that are formed in the fluid phase under physiologic conditions are rapidly inactivated by the fast-acting plasmin inhibitor, α2-antiplasmin. Plasminogen binds to fibrin and thus becomes incorporated in clots as they are produced; since plasmin that is formed when bound to fibrin is protected from α2-antiplasmin, it remains active. Activators of plasminogen of various types are found in most body tissues, and all cleave the same Arg-Val bond in plasminogen to produce the two-chain serine protease, plasmin (Figure 51–6). The specificity of plasmin for fibrin is another mechanism to regulate fibrinolysis. Via one of its kringle domains, plasmin (ogen) specifically binds lysine residues on fibrin and so is increasingly incorporated into the fibrin mesh as it cleaves it. (Kringle domains [Figure 51–2] are common protein motifs of about 100-amino-acid residues in length, that have a characteristic covalent structure defined by a pattern of three disulfide bonds.) Thus, the carboxypeptidase TAFIa (activated thrombin activatable fibrinolysis inhibitor) (Figure 51–7), which removes terminal lysines from fibrin, is able to inhibit fibrinolysis. Thrombin activates TAFI to TAFIa, thereby inhibiting fibrinolysis during clot formation.
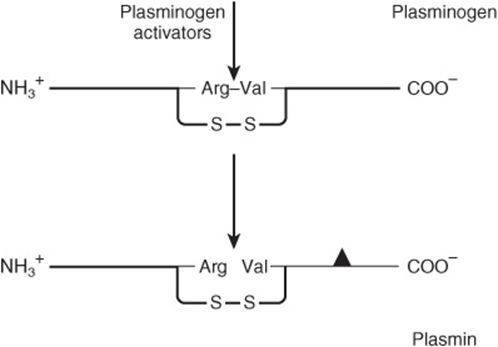
FIGURE 51–6 Activation of plasminogen. The same Arg-Val bond is cleaved by all plasminogen activators to give the two-chain plasmin molecule. The solid triangle indicates the serine residue of the active site. The two chains of plasmin are held together by a disulfide bridge.
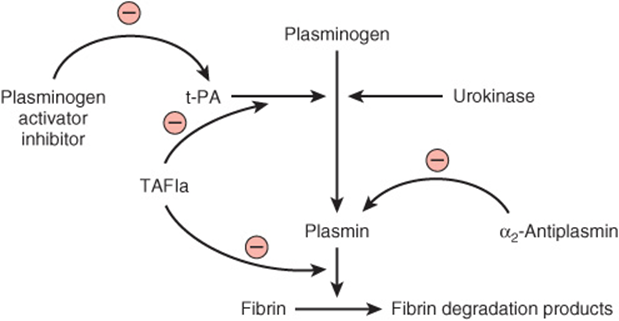
FIGURE 51–7 Initiation of fibrinolysis by the activation of plasmin. Scheme of sites of action of tissue plasminogen activator (t-PA), urokinase, plasminogen activator inhibitor, α2-antiplasmin, and thrombin-activatable fibrinolysis inhibitor (TAFIa) (the last three proteins exert inhibitory actions).
Tissue plasminogen activator (t-PA) (Figures 51-2 and 51-7) is a serine protease that is released into the circulation from vascular endothelium under conditions of injury or stress and is catalytically inactive unless bound to fibrin. Upon binding to fibrin, t-PA cleaves plasminogen within the clot to generate plasmin, which in turn digests the fibrin to form soluble degradation products and thus dissolves the clot. Neither plasmin nor the plasminogen activator can remain bound to these degradation products, and so they are released into the fluid phase, where they are inactivated by their natural inhibitors. Prourokinase is the precursor of a second activator of plasminogen, urokinase. Originally isolated from urine, it is now known to be synthesized by cell types such as monocytes and macrophages, fibroblasts, and epithelial cells. Its main action is probably in the degradation of extracellular matrix. Figure 51–7 indicates the sites of action of five proteins that influence the formation and action of plasmin.
Recombinant t-PA & Streptokinase Are Used as Clot Busters
Alteplase, t-PA produced by recombinant DNA technology, is used therapeutically as a fibrinolytic agent, as is streptokinase. However, the latter is less selective than t-PA, activating plasminogen in the fluid phase (where it can degrade circulating fibrinogen) as well as plasminogen that is bound to a fibrin clot. The amount of plasmin produced by therapeutic doses of streptokinase may exceed the capacity of the circulating α2-antiplasmin, causing fibrinogen as well as fibrin to be degraded and resulting in the bleeding often encountered during fibrinolytic therapy. Because of its relative selectivity for degrading fibrin, recombinant t-PA has been widely used to restore the patency of coronary arteries following thrombosis. If administered early enough, before irreversible damage of heart muscle occurs (about 6 h after onset of thrombosis), t-PA can significantly reduce the mortality rate from myocardial damage following coronary thrombosis. Streptokinase has also been widely used in the treatment of coronary thrombosis, but has the disadvantage of being antigenic.
t-PA has also been used in the treatment of ischemic stroke, peripheral arterial occlusion, and pulmonary embolism.
There are a number of disorders, including cancer and sepsis, in which the concentrations of plasminogen activators increase. In addition, the antiplasmin activities contributed by α1-antitrypsin and α2-antiplasmin may be impaired in diseases such as cirrhosis. Since certain bacterial products, such as streptokinase, are capable of activating plasminogen, they may be responsible for the diffuse hemorrhage sometimes observed in patients with disseminated bacterial infections.
Platelet Aggregation Requires Outside-In and Inside-Out Transmembrane Signaling
Platelets normally circulate in an unstimulated disk-shaped form. During hemostasis or thrombosis, they become activated and help form hemostatic plugs or thrombi. Three major steps are involved: (1) adhesion to exposed collagen in blood vessels, (2) release (exocytosis) of the contents of their storage granules, and (3) aggregation.
Platelets adhere to collagen via specific receptors on the platelet surface, including the glycoprotein complexes GPIa-IIa (α2β1 integrin; Chapter 52) and GPIb-IX-V, and GPVI. The binding of GPIb-IX-V to collagen is mediated via von Willebrand factor; this interaction is especially important in platelet adherence to the subendothelium under conditions of high shear stress that occur in small vessels and partially stenosed arteries.
Platelets adherent to collagen change shape and spread out on the subendothelium. They release the contents of their storage granules (the dense granules and the alpha granules); secretion is also stimulated by thrombin.
Thrombin, formed from the coagulation cascade, is the most potent activator of platelets and initiates activation by interacting with its receptors PAR (protease-activated receptor)-1, PAR-4, and GPIb-IX-V on the platelet plasma membrane (Figure 51–8A). The further events leading to platelet activation upon binding to PAR-1 and PAR-4 are examples of outside-in transmembrane signaling, in which a chemical messenger outside the cell generates effector molecules inside the cell. In this instance, thrombin acts as the external chemical messenger (stimulus or agonist). The interaction of thrombin with its G protein-coupled receptors PAR-1 and PAR-4 stimulates the activity of an intracellular phospholipase Cβ. This enzyme hydrolyzes the membrane phospholipid phosphati-dylinositol 4,5-bisphosphate (PIP2, a polyphosphoinositide) to form the two internal effector molecules, 1,2-diacylglycerol and 1,4,5-inositol trisphosphate.
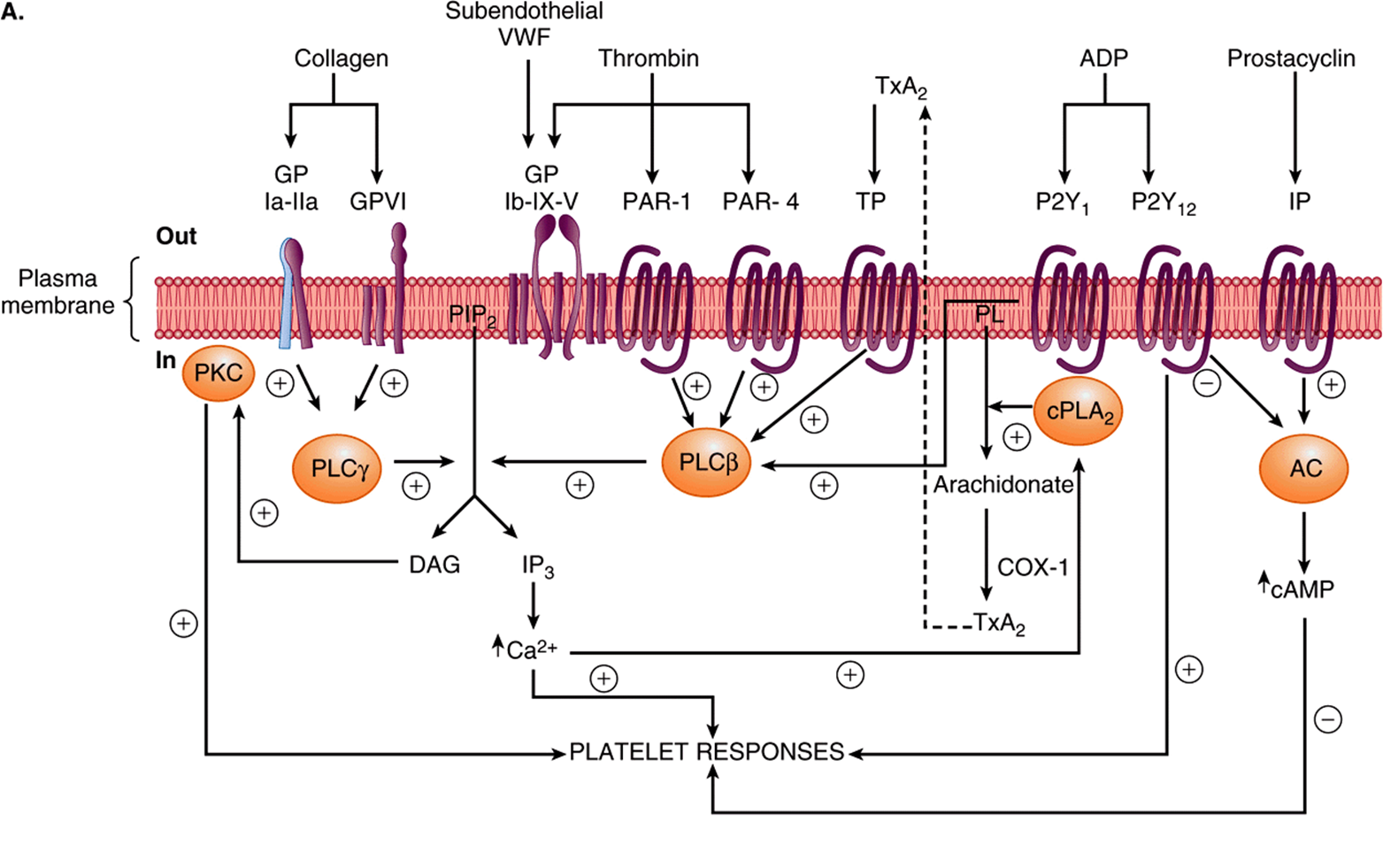
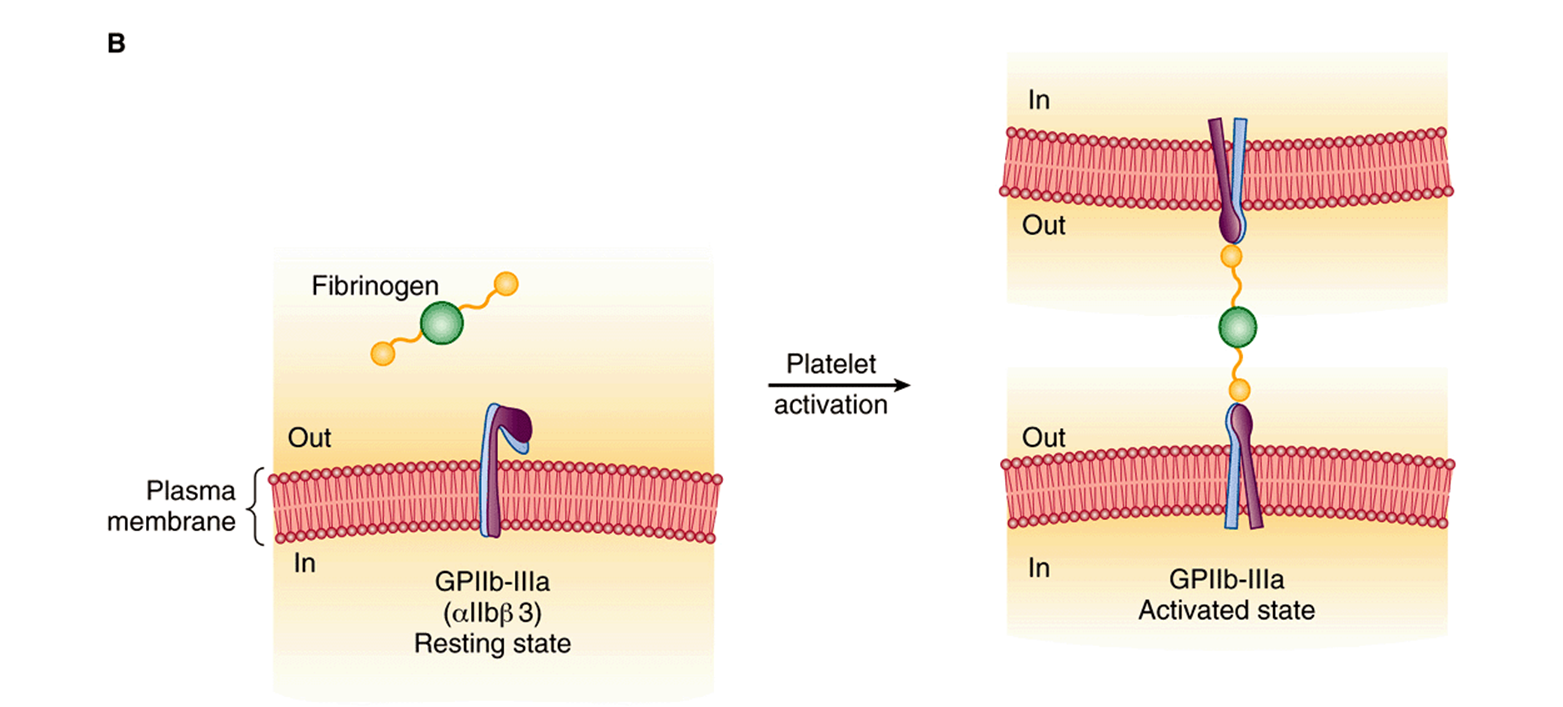
FIGURE 51–8 (A) Diagrammatic representation of platelet activation by collagen, thrombin, thromboxane A2 and ADP, and inhibition by prostacyclin. The external environment, the plasma membrane, and the inside of a platelet are depicted from top to bottom. Platelet responses include, depending on the agonist, change of platelet shape, release of the contents of the storage granules, and aggregation. (AC, adenylyl cyclase; cAMP, cyclic AMP; COX-1, cyclooxgenase-1; cPLA2, cytosolic phospholipase A2; DAG, 1,2-diacylglycerol; GP, glycoprotein; IP, prostacyclin receptor; IP3, inositol 1,4,5-trisphosphate; P2Y1, P2Y12, purinoceptors; PAR, protease activated receptor; PIP2, phosphatidylinositol 4,5-bisphosphate; PKC, protein kinase C; PL, phospholipid; PLCβ, phospholipase Cβ; PLCγ, phospholipase Cβ; TP, thromboxane A2 receptor; TxA2, thromboxane A2; VWF, von Willebrand factor.) The G proteins that are involved are not shown. (B) Diagrammatic representation of platelet aggregation mediated by fibrinogen binding to activated GPIIb-IIIa molecules on adajacent platelets. Signaling events initiated by all aggregating agents transform GPIIb-IIIa from its resting state to an activated form that can bind divalent fibrinogen or multivalent von Willebrand factor at the high shear that occurs in small vessels.
Hydrolysis of PIP2 is also involved in the action of many hormones and drugs. Diacylglycerol stimulates protein kinase C, which phosphorylates the protein pleckstrin (47 kDa). This results in aggregation and release of the contents of the storage granules. ADP released from dense granules can also activate platelets via its specific G protein-coupled receptors (Figure 51–8A), resulting in aggregation of additional platelets. IP3 causes release of Ca2+into the cytosol mainly from the dense tubular system (or residual smooth endoplasmic reticulum from the megakaryocyte), which then interacts with calmodulin and myosin light chain kinase, leading to phosphorylation of the light chains of myosin. These chains then interact with actin, causing changes of platelet shape.
Collagen-induced activation of a platelet cytosolic phospholipase A2 by increased levels of intracellular Ca2+ results in liberation of arachidonic acid from platelet membrane phospholipids, leading to the formation of thromboxane A2 (Chapter 23). Thromboxane A2, in turn, by binding to its G protein-coupled TP receptor, can further activate phospholipase C, promoting platelet aggregation (Figure 51–8A).
Activated platelets, besides forming a platelet aggregate, accelerate the activation of factor X and prothrombin by exposing the anionic phospholipid phosphatidylserine on their membrane surface (Figure 51–1).
All of the aggregating agents, including thrombin, collagen, ADP, and others such as platelet-activating factor, via an inside-out signaling pathway, modify the platelet surface glycoprotein complex GPIIb-IIIa (αIIbβ3; Chapter 52) so that the receptor has a higher affinity for fibrinogen or von Willebrand factor (Figure 51–8B). Molecules of divalent fibrinogen or multivalent von Willebrand factor then link adjacent activated platelets to each other, forming a platelet aggregate. von Willebrand factor-mediated platelet aggregation occurs under conditions of high shear stress. Some agents, including epinephrine, serotonin, and vasopressin, exert synergistic effects with other aggregating agents.
Endothelial Cells Synthesize Prostacyclin & Other Compounds That Affect Clotting & Thrombosis
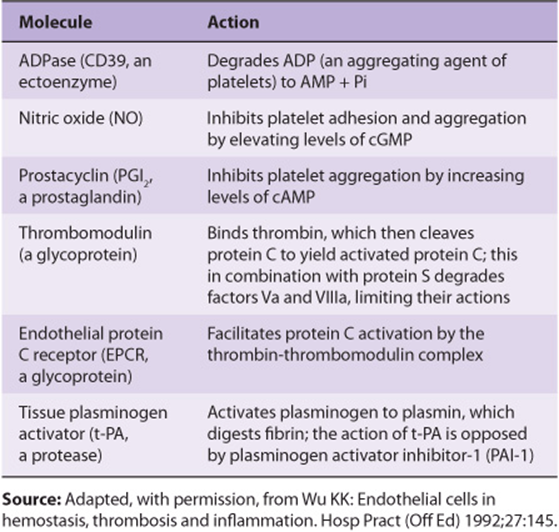
The endothelial cells in the walls of blood vessels make important contributions to the overall regulation of hemostasis and thrombosis. As described in Chapter 23, these cells synthesize the prostanoid prostacyclin (PGI2), a potent inhibitor of platelet aggregation. Prostacyclin acts by stimulating the activity of adenylyl cyclase in the surface membranes of platelets via its G protein-coupled receptor (Figure 51–8A). The resulting increase of intraplatelet cAMPopposes the increase in the level of intracellular Ca2+ produced by IP3 and thus inhibits platelet activation. This is in contrast with the effect of the prostanoid thromboxane A2 that is formed by activated platelets, which is that of promoting aggregation. Endothelial cells play other roles in the regulation of thrombosis. For instance, these cells possess an ADPase, which hydrolyzes ADP, and thus opposes its aggregating effect on platelets. In addition, these cells appear to synthesize heparan sulfate, an anticoagulant, and they also synthesize plasminogen activators, which may help dissolve thrombi. Table 51-3 lists some molecules produced by endothelial cells that affect thrombosis and fibrinolysis. Nitric oxide (endothelium-derived relaxing factor) is discussed in Chapter 49.
TABLE 51–3 Molecules Synthesized by Endothelial Cells That Play a Role in the Regulation of Thrombosis and Fibrinolysis
Analysis of the mechanisms of uptake of atherogenic lipoproteins, such as LDL, by endothelial, smooth muscle, and monocytic cells of arteries, along with detailed studies of how these lipoproteins damage such cells is a key area of study in elucidating the mechanisms of atherosclerosis (Chapter 26).
Aspirin Is One of Several Effective Antiplatelet Drugs
Certain drugs (antiplatelet drugs) inhibit platelet responses. The most commonly used antiplatelet drug is aspirin (acetylsalicylic acid), which irreversibly acetylates and thus inhibits the platelet cyclooxygenase system (COX-1) involved in formation of thromboxane A2 (Chapter 15), a potent aggregator of platelets and also a vasoconstrictor. Platelets are very sensitive to aspirin; as little as 30 mg/d (one regular aspirin tablet contains 325 mg) effectively eliminates the synthesis of thromboxane A2. Aspirin also inhibits production of prostacyclin (PGI2, which opposes platelet aggregation and is a vasodilator) by endothelial cells, but unlike platelets, these cells regenerate cyclooxygenase within a few hours. Thus, the overall balance between thromboxane A2 and prostacyclin can be shifted in favor of the latter, opposing platelet aggregation. Indications for treatment with aspirin include management of acute coronary syndromes (angina, myocardial infarction), acute stroke syndromes (transient ischemic attacks, acute stroke), severe carotid artery stenosis, and primary prevention of these and other atherothrombotic diseases.
Other antiplatelet drugs include clopidogrel, a specific inhibitor of the P2Y12 receptor for ADP, and antagonists of ligand binding to GPIIb-IIIa (eg, abciximab) that interfere with fibrinogen and von Willebrand factor binding and thus platelet aggregation.
Laboratory Tests Measure Coagulation, Thrombolysis, & Platelet Aggregation
A number of laboratory tests are available to measure the phases of hemostasis described above. The tests include platelet count, bleeding time/closure time, platelet aggregation, activated partial thromboplastin time (aPTT or PTT), prothrombin time (PT), thrombin time (TT), concentration of fibrinogen, fibrin clot stability, and measurement of fibrin degradation products. The platelet count quantitates the number of platelets. The skin bleeding time is an overall test of platelet and vessel wall function, while the closure time measured using the platelet function analyzer PFA-100 is an in vitro test of platelet-related hemostasis. Platelet aggregation measures responses to specific aggregating agents. aPTT is a measure of the intrinsic pathway and PT of the extrinsic pathway, with aPTT being used to monitor heparin therapy and PT, to measure the effectiveness of oral anticoagulants such as warfarin. The reader is referred to a textbook of hematology for a discussion of these tests.
SUMMARY
![]() Hemostasis and thrombosis are complex processes involving coagulation factors, platelets, and blood vessels.
Hemostasis and thrombosis are complex processes involving coagulation factors, platelets, and blood vessels.
![]() Many coagulation factors are zymogens of serine proteases, becoming activated, then inactivated during the overall process.
Many coagulation factors are zymogens of serine proteases, becoming activated, then inactivated during the overall process.
![]() Both extrinsic and intrinsic pathways of coagulation exist, the former initiated in vivo by tissue factor. The pathways converge at factor Xa, ultimately resulting in thrombin-catalyzed conversion of fibrinogen to fibrin, which is strengthened by covalent cross-linking, catalyzed by factor XIIIa.
Both extrinsic and intrinsic pathways of coagulation exist, the former initiated in vivo by tissue factor. The pathways converge at factor Xa, ultimately resulting in thrombin-catalyzed conversion of fibrinogen to fibrin, which is strengthened by covalent cross-linking, catalyzed by factor XIIIa.
![]() Genetic disorders that lead to bleeding occur; the principal disorders involve factor VIII (hemophilia A), factor IX (hemophilia B), and von Willebrand factor (von Willebrand disease).
Genetic disorders that lead to bleeding occur; the principal disorders involve factor VIII (hemophilia A), factor IX (hemophilia B), and von Willebrand factor (von Willebrand disease).
![]() Antithrombin is an important natural inhibitor of coagulation; genetic deficiency of this protein can result in thrombosis.
Antithrombin is an important natural inhibitor of coagulation; genetic deficiency of this protein can result in thrombosis.
![]() For their activity, factors II, VII, IX, and X and proteins C and S require vitamin K-dependent γ-carboxylation of certain glutamate residues, a process that is inhibited by the anticoagulant warfarin.
For their activity, factors II, VII, IX, and X and proteins C and S require vitamin K-dependent γ-carboxylation of certain glutamate residues, a process that is inhibited by the anticoagulant warfarin.
![]() Fibrin is dissolved by plasmin. Plasmin exists as an inactive precursor, plasminogen, which can be activated by tissue plasminogen activator (t-PA). Both t-PA and streptokinase are widely used to treat early thrombosis in the coronary arteries.
Fibrin is dissolved by plasmin. Plasmin exists as an inactive precursor, plasminogen, which can be activated by tissue plasminogen activator (t-PA). Both t-PA and streptokinase are widely used to treat early thrombosis in the coronary arteries.
![]() Thrombin and other agents cause platelet aggregation, which involves a variety of biochemical and morphologic events. Stimulation of phospholipase C and the polyphosphoinositide pathway is a key event in platelet activation, but other processes are also involved.
Thrombin and other agents cause platelet aggregation, which involves a variety of biochemical and morphologic events. Stimulation of phospholipase C and the polyphosphoinositide pathway is a key event in platelet activation, but other processes are also involved.
![]() Aspirin is an important antiplatelet drug that acts by inhibiting production of thromboxane A2.
Aspirin is an important antiplatelet drug that acts by inhibiting production of thromboxane A2.
REFERENCES
Colman RW, Marder VJ, Clowes AW, et al (editors): Hemostasis and Thrombosis: Basic Principles and Clinical Practice, 5th ed. Lippincott Williams & Wilkins, 2006.
Fauci AS, Braunwald E, Kasper DL, et al: Harrison’s Principles of Internal Medicine, 17th ed. McGraw-Hill, 2008.
Hoffman R, Benz Jr EJ, Shattil SJ, et al (editors): Hematology: Basic Principles and Practice, 4th ed. Elsevier Churchill Livingston, 2005.
Israels SJ (editor): Mechanisms in Hematology, 4th ed. Core Health Sciences Inc, 2011. (This text has many excellent illustrations of basic mechanisms in hematology.)
Michelson AD (editor): Platelets, 2nd ed. Elsevier, 2007.