CHEMICAL BIOLOGY
Cell Division, Small Molecules to Study
Michael A. Lampson and Tarun M. Kapoor, Rockefeller University, New York, New York
doi: 10.1002/9780470048672.wecb060
Cell division is the process by which a cell creates two genetically identical daughter cells. To maintain genomic integrity, a complex and highly regulated sequence of events ensures that the replicated chromosomes are partitioned equally between the daughter cells. For more than 50 years, strategies designed around small-molecule inhibitors have played a critical role in advancing our understanding of this essential process. Here we introduce a series of questions in the biology of cell division and illustrate how small molecules have been used to design experiments to address these questions. Because of the highly dynamic nature of cell division, the temporal control over protein function that is possible with small molecules has been valuable particularly in dissecting biologic mechanisms.
Biology of Cell Division
Cell division is the process by which a cell creates two genetically identical daughter cells. Each chromosome is replicated before mitosis begins, and after the division, the daughter cells inherit exactly one copy of each chromosome. A complex series of events have been divided broadly into two processes: mitosis, in which the identical sister chromosomes are separated and transported to opposite ends of the cell, and cytokinesis, in which the cell physically divides to create two daughter cells (Fig. 1). Preservation of genome integrity requires that the cell divide only after accurate segregation of sister chromosomes in mitosis. Failure in either chromosome segregation or in the timing of critical events in cell division leads to the loss or gain of whole chromosomes in the daughter cells, which is a condition known as aneuploidy that is strongly associated with developmental defects and human diseases such as cancer (reviewed in Reference 1). For over a century, cell division research has focused on mechanisms that physically segregate the chromosomes in mitosis and that divide the cell in cytokinesis, as well as those that control both processes in space and time. Some fundamental questions are how forces are generated to move chromosomes and to cleave the cell, how chromosome movements and cell cleavage are coordinated in space to ensure that each daughter cell inherits exactly one copy of each chromosome, and how events are coordinated in time so that chromosome segregation is complete before cell cleavage. Progress toward addressing these questions has integrated a structural picture of the machinery of cell division with an understanding of the molecular mechanisms that regulate that machinery.
____________________________
First published in Nature Chemical Biology 2, 19-27 (2006), doi:10.1038/nchembio757, © Nature Publishing Group, a division of Macmillan Publishers Limited. www.nature.com
Examining Cell Division with Small-Molecule Inhibitors
Our understanding of biologic processes such as cell division often develops from discovering or designing ways to perturb the process and observe the effects of the perturbation. Although genetic approaches have been used widely for this purpose, small-molecule inhibitors offer some distinct advantages. Small molecules provide a high degree of temporal control over protein function, generally acting within minutes or even seconds, and often are reversible, allowing both rapid inhibition and activation. The ability to design perturbations on short time scales has proven valuable particularly in examining dynamic biologic processes. All cell division takes place in approximately 1 hour, with many events on second-to-minute time scales, and several small-molecule inhibitors (Table 1) have played an integral role in dissecting the mechanisms of mitosis and cytokinesis.
The nature of spindle fibers
Progress through mitosis is linked closely to chromosome movements (Fig. 1a). Replicated chromosome pairs first move to the center of the cell. After all chromosomes are positioned correctly at metaphase (Fig. 1a iii), the sister chromosomes split apart at anaphase (Fig. 1a iv) and move to opposite sides of the cell before the cell divides into two daughter cells (Fig. 1a v, vi). Because of these coordinated movements, each daughter cell receives exactly one copy of each replicated chromosome. Correlated with chromosome movements is the appearance of a fibrous structure known as the mitotic spindle, initially observed in fixed samples, which forms at each mitosis and disappears after the chromosomes have separated. One great challenge in the study of cell division has been to understand the organization and function of the mitotic spindle, particularly in relation to chromosomes movements. The physical properties of the spindle fibers and how they might drive chromosome movements, as well as their molecular components, have been understood in part through use of the small molecule colchicine (Fig. 1b).
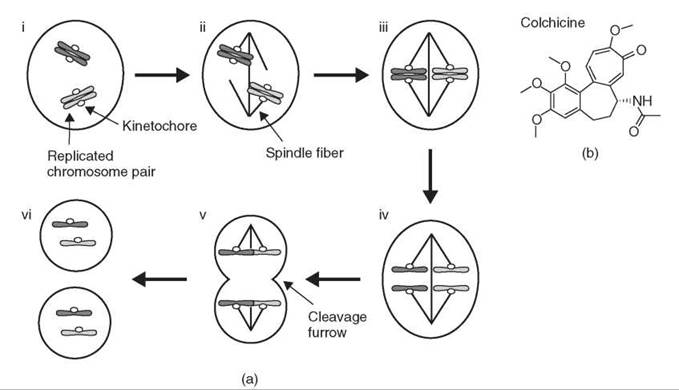
Figure 1. Overview of mitosis. (a) (i) Chromosomes are replicated before mitosis, and sister chromosomes are held together. (ii) The spindle forms and chromosomes attach to spindle fibers. (iii) Chromosomes move to the center of the spindle at metaphase. (iv) Sister chromosomes separate at anaphase and move in opposite directions. (v) The cell divides as the cleavage furrow forms between the separated chromosomes. (vi) Two daughter cells form, each with exactly one copy of each chromosome. (b) Structure of colchicine, a small molecule that targets microtubules.
Table 1. Small-molecule inhibitors used to study cell division
|
Small molecule |
Source |
Effects |
|
Colchicine |
Natural product from meadow saffron (genus Colchicum). |
Depolymerizes microtubules. |
|
Benomyl |
Synthetic, used as agricultural fungicide. |
Depolymerizes microtubules, effective in yeast. |
|
Nocodazole |
Synthetic, identified as antihelminthic compound (47). |
Depolymerizes microtubules. |
|
Taxol |
Natural product from bark of the yew tree (genus Taxus), identified for antitumor and antileukemic activity (56). |
Inhibits microtubule dynamics. |
|
Monastrol |
Synthetic, identified as cell-division inhibitor (24). |
Inhibits the kinesin-5 motor protein. |
|
Hesperadin |
Synthetic, identified as cell-division inhibitor (36). |
Inhibits Aurora kinases. |
|
AKI-1 |
Synthetic, identified as kinase inhibitor (57). |
Inhibits Aurora kinases. |
|
Cytochalasin B |
Natural product from the fungus Helminthosporium dematioideum (58). |
Depolymerizes actin filaments. |
Exploiting the fact that the spindle fibers are optically anisotropic, or birefringent, with different indices of refraction in different directions (i.e., parallel or perpendicular to the fiber axis), Inoue (2) developed a sensitive polarized light microscope to visualize directly the fibers in a living cell (2). He used this method to examine the behavior of the fibers after exposure to colchicines, a small molecule that was known to disrupt spindle function. Colchicine eliminated the birefringence over a time course that ranged from a few minutes to an hour depending on the concentration, which indicates a loss of the fibers (3). After the removal of colchicine, the fibers recovered to their original state. The inhibition of protein synthesis during the recovery demonstrated that the fibers were assembled from an available pool of material rather than by synthesis of new proteins (4). These findings, together with similar results obtained by chilling cells to eliminate the fibers (5), suggested that the spindle fibers consist of oriented polymers (hence the birefringence) in equilibrium with free molecules in solution. Treatment with colchicine or chilling pushes the equilibrium toward the depolymerized state, whereas the removal of colchicine or rewarming allows the system to return to its original state.
The same experimental paradigm, which combines the observation of spindle fibers with the perturbation of their function in living cells, was used to demonstrate the potential functional significance of the spindle fiber dynamics. When spindles were treated with low concentrations of colchicine, which did not eliminate the birefringence immediately, the fibers slowly contracted and pulled the chromosomes toward one pole of the spindle, which was anchored at the cell surface. After the removal of colchicine, the fibers elongated as chromosomes moved away from the pole (3, 6). This finding demonstrated that polymerization and depolymerization of the fibers could generate force by coupling to chromosome movement. These experiments exploited both the reversibility of colchicine and the ability to manipulate the behavior of the fibers by controlling the compound concentration, both properties of many small molecules.
In the Inoue studies, colchicine was used as an experimental tool to probe spindle function, but its target and mechanism of action were unknown. As 100 nM colchicine was sufficient to arrest cultured cells in mitosis, the implied tight binding suggested that the inhibitor might be used to isolate a colchicine-protein complex. A breakthrough came when colchicine was labeled with H3 with high specific activity to demonstrate reversible binding to cellular sites (7). When the labeled colchicine was tested with a variety of cells, tissues, and organelles, high-binding activity was observed with the mitotic spindle, cilia, sperm tails, and brain tissue (8, 9). A common feature of these preparations is that all are enriched in intracellular fibers called microtubules, the same fibers observed by Inoue, which suggested that the target of colchicine was a subunit of microtubules. The colchicine-binding activity was extracted by dissolving isolated sea urchin sperm tails, purified by gel filtration and sedimented over a sucrose gradient to identify a single component 10. The same component was isolated from mammalian brain, shown to bind GTP, and biochemically characterized as a GTP-binding protein (11). The protein was named tubulin because it was believed to be the primary constituent of microtubules (12).
It has been known for over 100 years that treatment with colchicine arrests cells in mitosis (reviewed in Reference 13). Other small molecules since then have been identified that also block mitosis by targeting microtubules. The potential of these compounds as cancer therapeutics was demonstrated by the vinca alkaloids, such as vincristine and vinblastine, which have been used in the clinic for 40 years. At high concentrations (10-100 nM), these compounds depolymerize microtubules, which eliminates the mitotic spindle. At lower concentrations that are used clinically, microtubules remain stable but microtubule dynamics are suppressed. Another small molecule that inhibits microtubule dynamics, taxol, also arrests cells in mitosis and is used widely to treat a variety of cancers (reviewed in Reference 14). The mitotic arrest induced by these drugs eventually leads to cell death (15) through mechanisms that are only beginning to be understood (16-18).
Progression Through Mitosis
It is clear from observing chromosome movements that cell division occurs in an ordered sequence of events (Fig. 1a). First chromosomes attach to spindle microtubule fibers and move to the spindle equator. Only after completion of this step do sister chromosomes separate at anaphase and move to opposite sides of the cell, followed by their division into two daughter cells. Events must occur in this order for successful chromosome segregation. If the cell enters anaphase prematurely, before chromosomes have attached properly to the spindle, the sister chromosomes will not segregate equally, which leads to aneuploid daughter cells. Therefore, mechanisms that determine the timing of anaphase onset are critical for the success of mitosis.
One hypothesis for how anaphase onset might be regulated was through feedback control as a mechanism of regulating passage through the cell cycle. At a specific point, known as a checkpoint, completion of an event generates a signal that allows the cell cycle to progress. Failure to complete the event causes a cell cycle arrest. Feedback control can be demonstrated experimentally by creating conditions, such as genetic mutations, under which the cell cycle arrest is bypassed. This strategy was used to show that a feedback control mechanism makes entry into mitosis dependent on the completion of DNA replication (19, 20). In the context of progression through mitosis, a critical process such as spindle assembly would be monitored to generate a signal that regulates anaphase onset. The mitotic arrest induced by colchicine, which disrupts the spindle by depolymerizing microtubules, is consistent with such a feedback mechanism. However, direct inhibition of a microtubule-dependent process required for anaphase also might explain the effect of colchicine. A prediction of the feedback control hypothesis is that mutations in genes required for feedback signaling would allow cells to bypass the colchicine-induced arrest and progress through mitosis without completing spindle assembly.
A genetic screen was devised to identify such mutations in budding yeast by using benomyl, a small-molecule inhibitor of microtubule polymerization that is effective in yeast, to perturb spindle assembly. The advantage of using a small molecule was that it could either be washed out, as the effect is reversible, or used at a low dose, so that cells would survive the treatment. With high benomyl (70 μg/ml), which prevents spindle formation, cells arrested in mitosis (Fig. 2a). After the removal of benomyl, cells recovered, proceeded normally through mitosis, and continued to grow (Fig. 2b) (21). Alternatively, with low benomyl (15 μg/ml), spindle assembly was slowed and mitotic exit was delayed to allow the completion of spindle assembly, but cells continued to grow (22). In both cases, cells defective in feedback control were expected to enter anaphase prematurely in the presence of benomyl with incomplete or nonexistent spindles, which would lead to massive chromosome missegregation and cell death (Fig. 2c, d). The difference in survival between cells with functional and defective feedback control provided a phenotype that could be used to screen for mutations in genes required for feedback control (21, 22). Cells were mutagenized, and mutants that failed to grow after benomyl treatment were selected (Fig. 2f). As in the Inoue studies with colchine, the reversibility of the small molecule and the ability to achieve partial inhibition by decreasing the dose were important components of the benomyl screening strategies.
Genes identified in the benomyl screens were named mad, for mitotic arrest deficient, or bub, for budding uninhibited by benomyl. The existence of these genes provided evidence for a feedback mechanism that delays anaphase onset until completion of spindle assembly, now often referred to as the mitotic spindle checkpoint. Much of our understanding of mitotic checkpoint signaling has developed from the mad and bub genes identified in the benomyl screens, which generally are well conserved from yeast to mammals. The importance of several of these genes for faithful chromosome segregation has been confirmed in transgenic mice, in which reduced expression increases both aneuploidy and cancer susceptibility, and human tumor cells have been reported to carry mutations in Mad1, Mad2, Bub1, and BubR1, a related vertebrate protein (reviewed in Reference 1). Additionally, human germline mutations in BubR1 have been linked to mosaic variegated aneuploidy, a condition associated with a high risk of cancer (23).
Benomyl was used in the mad and bub screens not because of its specific protein target but because of the perturbation of spindle assembly. In principle, the same experiments could have been done without knowing the protein target or by targeting a different component of the spindle. The generality of the spindle checkpoint has been demonstrated through the use of monastrol, a small-molecule inhibitor of the mitotic kinesin Eg5, which was identified in a screen for small molecules that arrest cells in mitosis without targeting tubulin (24). Monastrol treatment arrests cells in mitosis with monopolar spindles because Eg5 is required to separate the spindle poles. The inhibition of Mad2 by the microinjection of inhibitory antibodies overrides the checkpoint so that cells enter anaphase in the presence of monastrol with monopolar spindles (25). This finding indicates that the principle of feedback control generally applies to spindle perturbations through highly conserved mechanisms.
Inhibitors of Eg5 currently are in development as anticancer drugs because, like taxol and the vinca alkaloids, they arrest cells in mitosis by activating the spindle checkpoint. Recent studies have demonstrated that the efficacy of drugs targeting either Eg5 or microtubules requires a prolonged, checkpoint-dependent mitotic arrest (17, 26). A compromised spindle checkpoint, for example through reduced expression of Mad2, confers resistance to these drugs.

Figure 2. Screening strategy used to identify genes required for feedback control of anaphase onset in budding yeast (21). (a-d) Wild-type cells treated with benomyl arrest in mitosis (a), whereas mutant cells fail to arrest and enter anaphase without forming a spindle, which leads to chromosome missegregation (c). After the removal of benomyl, wild-type cells form a spindle, proceed through mitosis, and grow normally (b), but mutants do not survive (d). (e) Structure of benomyl. (f) To screen for mutations in genes required for feedback control, cells were mutagenized, and colonies grown from single cells were transferred to create two replicate plates. One plate (i) was grown without benomyl. The second plate (ii) was treated with benomyl. Colonies that failed to grow on the second plate, which indicates defective feedback control, were selected from the first plate to identify the mutated gene.
Primary signals for checkpoint activation: attachment or tension?
Whereas the benomyl screens established the existence of the spindle checkpoint and identified some of the key components in checkpoint signaling, a fundamental question that remained unanswered was what exactly is monitored by the checkpoint. Two general models have been proposed. One is that the checkpoint monitors the attachment of spindle microtubules at the kinetochore, a structure that forms on each chromosome to mediate microtubule binding. Unattached kinetochores keep the checkpoint active and delay anaphase (27). A second model is that the checkpoint monitors force across the centromere, the region of the chromosome where kinetochores assemble (28). When both sister kinetochores are attached correctly, they are pulled in opposite directions by the microtubule fibers and the centromere is under tension (Fig. 3a). In this model, the absence of centromere tension would keep the checkpoint active. Small molecules that target tubulin have provided a way to test these models experimentally. Nocodazole depolymerizes microtubules, which creates unattached kinetochores (Fig. 3c), whereas taxol stabilizes microtubules but inhibits their dynamics, which decreases the centromere tension (Fig. 3b) (29).
To determine the effects of these microtubule perturbations on spindle checkpoint signaling, intracellular localization of Mad2 was examined. At early stages of mitosis (Fig. 1a i, ii), Mad2 localizes to kinetochores. As cells progress through mitosis, however, Mad2 disappears from kinetochores, and at anaphase onset (Fig. 1a iv) none of the kinetochores have detectable Mad2. These findings suggest that the presence of Mad2 on kinetochores serves as a signal to delay anaphase (29-31).
When microtubules are depolymerized with nocodazole (Fig. 3c), Mad2 localizes to all kinetochores, which indicates the activation of the checkpoint. If microtubule dynamics are suppressed with taxol while maintaining kinetochore attachments (Fig. 3b), Mad2 localizes to only a few kinetochores (29). This finding suggests that checkpoint signaling, as determined by Mad2 localization, is sensitive to attachment but does not respond directly to centromere tension. The interpretation of these experiments is complicated, however, because tension is required for kinetochores to bind the full complement of microtubules; loss of tension may activate the checkpoint indirectly (32).
Experiments in yeast suggested that a member the Aurora kinase family, Ipl1, is required to activate the spindle checkpoint in response to loss of tension but not loss of microtubule attachments (33). Understanding the function of Aurora kinases is particularly important because they have been linked to oncogenesis, and Aurora kinase inhibitors currently are in development as cancer therapeutics (34, 35). In mammalian cells, the inhibition of Aurora kinase activity with small-molecule inhibitors has been shown to bypass the mitotic arrest induced by taxol but not that induced by nocodazole, which is consistent with the idea that the kinase is required specifically in a tension-sensitive mechanism of checkpoint activation (36, 37). The interpretation of these results is complicated, however, because Aurora kinases are implicated also in regulating kinetochore-microtubule binding (38, 39). An alternative interpretation is that Aurora kinase inhibition overrides the taxol-induced arrest through effects on kinetochore-microtubule attachments.

Figure 3. Manipulation of chromosome-microtubule attachments with small molecules. (a) In the absence of microtubule poisons, the attachment of both kinetochores to spindle microtubules creates tension across the centromere. (b) Taxol reduces tension across the centromere by inhibiting microtubule dynamics. (c) Nocodazole creates unattached kinetochores by depolymerizing microtubules.
Correcting errors in chromosome spindle attachments
Feedback control of anaphase onset, or mitotic checkpoint signaling, is one mechanism that contributes to ensuring accurate chromosome segregation. Delaying anaphase in response to unattached kinetochores, however, is not sufficient. Chromosomes must attach to spindle microtubules in a particular orientation. For each replicated chromosome pair, the sister kinetochores attach to opposite poles of the spindle so that when sister chromosomes separate at anaphase, they are pulled to opposite sides of the cell. Other attachment states can occur, for example if both sister kinetochores are attached to the same spindle pole or a single kinetochore is attached to both poles. If these errors are not corrected, sister chromosomes will not segregate properly at anaphase (40).
Error correction is thought to occur by stabilizing correct attachments while destabilizing incorrect attachments (41). Experiments in yeast showed that the inhibition of the Ipl1/Aurora family of kinases prevents error correction by stabilizing incorrect attachments (38, 42), but how the active kinase corrected attachment errors was not known. This problem was particularly difficult to address because attachment errors are observed infrequently in the presence of active Aurora kinase (43). Experimental approaches that accumulated attachment errors through inhibition of Aurora kinase, for example by genetic mutation (42), did not permit subsequent kinase activation to examine error correction. Reversible small-molecule Aurora kinase inhibitors present a solution to this problem because they can be used to inhibit kinase function and subsequently removed to activate the kinase.
To devise a strategy to address the question of how attachment errors were corrected, several issues needed to be addressed. First, Aurora kinases have been implicated in multiple processes in mitosis (44). Ideally, kinase inhibition temporally would be controlled to isolate experimentally the error correction process. Second, the microtubules fibers that attach chromosomes to the spindle are highly dynamic, and the error correction likely involves some regulation of these dynamics. Live imaging would permit the analysis of microtubule dynamics with high temporal and spatial resolution. Finally, analysis of microtubule dynamics is difficult if individual fibers are obscured by other microtubules in the spindle. By creating conditions in which the improperly attached chromosomes are positioned away from the spindle body, individual fibers could be observed clearly.
An assay was developed, using several small-molecule inhibitors, in which all of these issues were addressed (Fig. 4a) (45). First cells were arrested in mitosis with monopolar spindles using the Eg5 inhibitor monastrol (Fig. 4a i). In the monopolar spindles many chromosomes have a particular attachment error in which both sisters are attached to the single spindle pole, referred to as syntelic attachment (46). After the removal of monastrol, the spindle becomes bipolar, all attachment errors are corrected, and the chromosomes segregate normally at anaphase. To test if Aurora kinase activity is required for correction of the attachment errors, an Aurora kinase inhibitor was added immediately after the removal of monastrol (Fig. 4a ii). The advantage of adding the Aurora kinase inhibitor at this point is that Aurora kinase activity is unperturbed for all the preceding stages of mitosis. This assay was performed with two structurally unrelated Aurora kinase inhibitors, AKI-1 and hesperadin, to control for possible off-target activities of the small molecules (Fig. 4b).
Using cells that express GFP-labeled tubulin, both chromosome and microtubule dynamics were examined at high resolution by multimode fluorescence and transmitted light microscopy during spindle bipolarization in the presence of the Aurora kinase inhibitor. The syntelic attachment errors persisted as the spindle bipolarized, which directly demonstrates that Aurora kinase activity is required for the correction of these errors. Notably, some of the improperly attached chromosomes were positioned away from the spindle body, which allowed clear observation of the attached microtubule fiber, unobstructed by other spindle microtubules (Fig. 4c). At this point, the Aurora kinase inhibitor was removed to examine how the active kinase might correct the syntelic attachment errors (Fig. 4a iii, iv). Aurora kinase was shown to be fully active in vivo 30-60 minutes after removing the inhibitor, as determined by measuring the phosphorylation of histone H3, a known Aurora kinase substrate. On the time scale of kinase activation, improperly attached chromosomes remained attached to the microtubule fibers and were pulled to the spindle pole as the fibers shortened (Fig. 4d). After disassembly of the microtubule fibers, the chromosomes moved to their usual position at the center of the spindle as correct attachments formed. Properly attached chromosomes were not affected, which suggests the local regulation of microtubule dynamics by Aurora kinase activity.
This assay demonstrates some advantages of small-molecule inhibitors, particularly in combination with high-resolution live-cell microscopy. Mitosis is a highly dynamic process with many events occurring on time scales of minutes or seconds. Ideally, an experiment would allow both perturbation of protein function and observation of the effects of the perturbation on similar time scales. The use of reversible small-molecule inhibitors to manipulate protein function, together with live cell imaging, makes this possible. In the assay described here, the inhibitors were used effectively as switches to turn enzymes on and off for both the kinesin Eg5 and Aurora kinases. With this high degree of temporal control, a mechanism for correcting chromosome attachment errors could be dissected without perturbing the preceding processes, such as those involved in spindle assembly.

Figure 4. Correction of improper chromosome attachments by activation of Aurora kinase (45). (a) Assay schematic. (i) Treatment with the Eg5 inhibitor monastrol arrests cells in mitosis with monopolar spindles, in which sister chromosomes often are both attached to the single spindle pole. (ii) Hesperadin, an Aurora kinase inhibitor, is added as monastrol is removed. As the spindle bipolarizes with Aurora kinase inhibited, attachment errors fail to correct so that some sister chromosomes are still attached to the same pole of the bipolar spindle. (iii) Removal of hesperadin activates Aurora kinase. Incorrect attachments are destabilized by disassembling the microtubule fibers, which pulls the chromosomes to the pole, whereas correct attachments are stable. (iv) Chromosomes move from the pole to the center of the spindle as correct attachments form. (b) Structures of the Eg5 inhibitor monastrol and two Aurora kinase inhibitors, hesperadin and AKI-1. (c) Spindles were fixed after bipolarization either in the absence (i) or presence (ii) of an Aurora kinase inhibitor. Arrows indicate sister chromosomes that are both attached to the same spindle pole. Projections of multiple image planes are shown, with optical sections of boxed regions (1 and 2) to highlight attachment errors. Scale bars 5 μm. (d) After the removal of hesperadin, GFP-tubulin (top) and chromosomes (bottom) were imaged live by three-dimensional confocal fluorescence microcopy and DIC, respectively. Arrow and arrowhead show two chromosomes that move to the spindle pole (marked by circle in DIC images) as the associated kinetochore-microtubule fibers shorten and that then move to the center of the spindle. Time (minutes:seconds) after the removal of hesperadin. Scale bar 5 μm.
Force generation in cytokinesis
Following inactivation of the checkpoint, the chromosomes segregate at anaphase and are pulled in opposite directions by the attached microtubule fibers. After anaphase, the cell physically divides into two daughter cells in cytokinesis. Several key questions in cytokinesis have been addressed by using small-molecule inhibitors. First, how is the force generated to cleave the cell into two parts? Second, what mechanisms determine the position of the cleavage plane? Third, how is the timing of progression through cytokinesis controlled?
Cleavage of a cell into two equal parts is a dramatic event that requires coordinated generation of force around the entire perimeter of the cell (Fig. 5a). As a clue to how this might be achieved, a filamentous structure was shown by electron microscopy to lie at the cleavage furrow, just below the plasma membrane (reviewed in Reference 47). These filaments, distinct from microtubules, were called microfilaments. A key step in understanding the function of microfilaments in cytokinesis and other processes was to observe a correlation between the presence of the filaments, their disruption by the small molecule cytochalasin (Fig. 5b), and the phenotype of cytochalasin treatment. Cytochalasin eliminated the microfilaments at the cleavage furrow and prevented contraction of the furrow at cytokinesis. Cytochalasin also inhibited several other forms of cellular or intracellular force generation, including cell motility, membrane ruffling, and nerve outgrowth (48, 49). Microfilaments were observed in all of these systems, and in every case the microfilaments were disrupted by cytochalasin and returned to their normal state as cells recovered after the removal of cytochalasin. Furthermore, the actions of cytochalasin and colchicine generally were mutually exclusive: Processes dependent on microtubules and therefore inhibited by colchicine were often insensitive to cytochalasin, whereas those inhibited by cytochalasin were generally insensitive to colchicine (49). These observations suggested that the two types of filamentous structures could function independently in the cell. Although the molecular target of cytochalasin was still unknown, the correlative evidence indicated that microfilaments played a fundamental role in the generation of forces at the cellular level: “[T]he evidence seems overwhelming that microfilaments are the contractile machinery of nonmuscle cells” (49). Contractility in muscle was achieved through the action of the myosin motor, which uses energy from ATP hydrolysis to slide filaments made up of polymers of the protein actin. The relevance of this process to other cell types had not been demonstrated.
A direct link between cytochalasin and actin was provided by the demonstration that cytochalasin decreases the viscosity of actin filaments purified from muscle (50). This experiment led to two important conclusions. First, cytochalasin interacts directly with actin. Second, “an interaction of cytochalasin with actin or actin-like proteins in vivo could account for the ability of cytochalasin to inhibit various forms of cell motility and contraction” (50). Thus, actin was shown to be the molecular target of cytochalasin and implicated as a critical component of the microfilaments involved in cytochalasin-sensitive processes, including contraction of the cleavage furrow at cytokinesis.
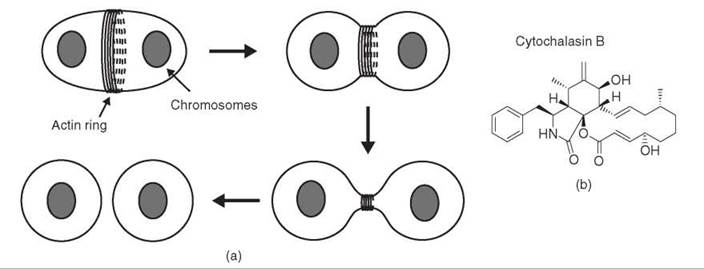
Figure 5. Force production by the contractile ring in cytokinesis. (a) A ring of actin filaments forms at the plasma membrane and contracts to divide the cell in half. (b) Structure of cytochalasin B, a small molecule that targets actin.
Spatial and temporal control of cytokinesis
The cleavage plane typically is positioned in the center of the cell so that cellular components are divided equally between the two daughter cells. Asymmetric divisions do occur, however, and are particularly important during development, when the location of the cleavage plane can determine the fate of the daughter cells. Models to explain the position of the cleavage plane relied on the presence of the bipolar microtubule array of the mitotic spindle, which would place the division plane in between the spindle poles. To test this idea directly, an experiment was designed using monastrol to determine if cytokinesis could occur in cells with monopolar spindles (25). Because the spindle checkpoint prevents anaphase onset in monastrol-arrested cells, inhibitory antibodies against Mad2 were microinjected to override the checkpoint. The microinjected cells entered anaphase and successfully completed cytokinesis (Fig. 6). This experiment demonstrated that a bipolar microtubule array is not required for cytokinesis. Furthermore, careful analysis of microtubule dynamics showed that a population of microtubules near the chromosomes was stabilized during anaphase in the monopolar spindles at the location where the cleavage plane formed. These findings suggested a model in which microtubule dynamics are regulated through association with chromosomes to determine the position of the division plane.

Figure 6. Assay to examine cytokinesis in the presence of a monopolar spindle (25). Treatment with monastrol, a small-molecule inhibitor of the kinesin Eg5, causes cells to arrest in mitosis with monopolar spindles because of activation of the spindle checkpoint. Microinjection of an antibody against the protein Mad2 inactivates the checkpoint so that cells divide with monopolar spindles.
The monastrol experiment showed that the positioning of the cleavage plane is correlated with the position of a particular population of microtubules. How microtubules generate a signal to recruit components of the contractile machinery remained an outstanding question. It has been difficult to isolate experimentally the molecular events that occur in the short time between anaphase onset and the beginning of cytokinesis. To address this problem, a strategy was devised to arrest cytokinesis at a defined point, before contraction of the cleavage furrow (51). A small-molecule inhibitor of the ATPase activity of nonmuscle myosin II, the actin-based motor that generates the force to contract the cleavage furrow, was identified in a high-throughput screen. This inhibitor, called blebbistatin because it prevents myosin II-dependent membrane blebbing, blocks cytokinesis with components of the cleavage furrow such as myosin II itself assembled in the correct position, but without any contraction of the furrow. With cytokinesis arrested at this point, other small-molecule inhibitors were used to dissect the molecular requirements for furrow positioning. The advantage of adding inhibitors during the blebbistatin arrest is that their effects on a single process could be isolated without affecting the preceding processes. These experiments showed that signals from both Aurora and Rho kinases are required to localize myosin II to the cleavage furrow.
To investigate the timing of cytokinesis, small molecules have been used in several strategies to perturb the cleavage process. If cytokinesis is prevented by perturbing either actin or microtubules with cytochalasin or nocodazole, or by inhibiting myosin II with blebbistatin, a window of approximately 1 hour exists during which cytokinesis can occur if the inhibitor is removed (51-53). The existence of this window suggests that an irreversible step exists that prevents cells from reversing progress through the cell cycle and returning to cytokinesis. Such irreversible steps can be mediated by the degradation of key proteins through proteolysis. To test whether proteolysis is required for the irreversible exit from cytokinesis, the small-molecule proteasome inhibitor MG132 was added during the blebbistatin arrest. In the presence of MG132, the time that cells remained in cytokinesis, as determined by the presence of myosin II at the cleavage furrow, increased substantially (51). This experiment demonstrated that the duration of cytokinesis is determined by ubiquitin-mediated proteolysis.
Conclusion
The experiments described in this review illustrate how small-molecule inhibitors have been used to design strategies to address fundamental questions in cell division. As our understanding of cell division advances, the use of small molecules should complement genetic and RNAi-based approaches. In particular, the temporal control over protein function that is possible with small molecules makes it possible to dissect the functions of proteins that are involved in multiple processes. Part of the complexity of cell division is that many proteins, for example the Aurora and Polo family kinases (44, 54), are implicated at multiple stages in both mitosis and cytokinesis. The perturbation of one stage often affects subsequent events, such as by activation of the spindle checkpoint, which limits analyses by preventing subsequent steps. Small-molecule inhibitors can be used to temporally isolate a specific process without perturbing the preceding events. These strategies will likely make important contributions to future investigations of cell division mechanisms.
It also is important to consider some of the limitations of small-molecule inhibitors, particularly in comparison to genetic approaches. With genetics, any gene can be targeted for mutation or deletion without directly affecting any other gene. The discovery of useful small-molecule inhibitors, however, is challenging. The specificity of small-molecule inhibitors also is difficult to demonstrate convincingly. Testing a kinase inhibitor against over 500 kinases in the human genome, for example, is a substantial undertaking. One way to address specificity is to use small molecules in focused assays, in which a narrowly defined biologic process is examined, so that off-target effects are unlikely to be relevant. In combination with this approach, the effects of different, chemically unrelated inhibitors that target the same protein could be compared, as the off-target activities are unlikely to be similar.
Future directions
The use of small-molecule inhibitors is limited only by the availability of inhibitors and the assays that can be designed around them. The proteins that currently are known to be targeted by small molecules make up a small fraction of the proteome. The identification of new inhibitors will promote the application of small molecule-based strategies to an increasing range of biologic problems. As methods are developed to monitor protein function with high temporal and spatial resolution, particularly in living cells, the scope for using small molecules also will increase. With recent advances in fluorescence-based probes, it has become possible to monitor numerous properties of living cells, including protease activity, posttranslational modifications, membrane potential, and pH, as well as mediators of intracellular signaling such as Ca2+ and cyclic AMP (55). The temporal control available with small-molecule inhibitors, combined with these high-resolution readouts, should be a powerful combination for examining dynamic biologic processes in living cells. In vitro methods also have been developed for measuring the enzymatic activities of single protein molecules. Observing the effects of a small-molecule inhibitor both at this level and in a more complex cellular context should provide new insights into protein function.
Both the identification of new inhibitors and the design of increasingly sophisticated assays to examine their effects also will contribute to the drug discovery process in several ways. First, studies with small-molecule inhibitors will advance our understanding of the effects of chemical inhibition, the mode of action of most drugs, which typically does not affect levels of the target protein. Approaches such as RNAi that act by reducing protein levels may have other effects, for example, preventing the formation of a multiprotein complex for which the depleted protein is required. Second, the discovery of new inhibitors may provide leads for drug development. For example, inhibitors that induce a mitotic arrest by targeting a protein that is specific to mitosis are potential leads for anticancer drugs that will have fewer side effects than tubulin poisons such as taxol and the vinca alkaloids. Third, assays that are designed to examine the effects of small molecules on specific cellular processes can be used to screen potential drugs for both on- and off-target activities. Finally, a deeper understanding of the basic molecular mechanisms of key cellular processes should lead to improved therapeutic strategies.
References
1. Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 2005; 5:773-85.
2. Inoue S. Polarization optical studies of the mitotic spindle. I. The demonstration of spindle fibers in living cells. Chromosoma 1953; 5:487-500.
3. Inoue S. The effect of colchicine on the microscopic and submicroscopic structure of the mitotic spindle. Exp. Cell Res. 1952; 2:305-318.
4. Inoue S, Sato H. Cell motility by labile association of molecules. The nature of mitotic spindle fibers and their role in chromosome movement. J. Gen. Physiol. 1967; 50:259-292.
5. Inoue S. Organization and Function of the Mitotic Spindle, in Primitive Motile Systems. In: Cell Biology. Allen RD, Kamiya K, eds. 1964. Academic, New York.
6. Inoue S. Cell division and the mitotic spindle. J. Cell. Biol. 1981; 91:131-147.
7. Taylor EW. The Mechanism of Colchicine Inhibition of Mitosis. I. Kinetics of Inhibition and the Binding of H3-Colchicine. J. Cell. Biol. 1965; 25:145-160.
8. Borisy GG, Taylor EW. The mechanism of action of colchicine. Binding of colchincine-3H to cellular protein. J. Cell. Biol. 1967; 34:525-533.
9. Borisy GG, Taylor EW. The mechanism of action of colchicine Colchicine binding to sea urchin eggs and the mitotic apparatus. J. Cell. Biol. 1967; 34:535-548.
10. Shelanski ML, Taylor EW. Isolation of a protein subunit from microtubules. J. Cell. Biol. 1967; 34:549-554.
11. Weisenberg RC, Borisy GG, Taylor EW. The colchicine-binding protein of mammalian brain and its relation to microtubules. Biochemistry 1968; 7:4466-4479.
12. Mohri H. Amino-acid composition of “Tubulin” constituting microtubules of sperm flagella. Nature 1968; 217:1053-1054.
13. Mitchison TJ, Salmon ED. Mitosis: a history of division. Nat. Cell. Biol. 2001; 3:17-21.
14. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004; 4:253-265.
15. Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L. Mitotic block induced in HeLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res. 1996; 56:816-825.
16. Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell 2005; 8:7-12.
17. Tao W, South VJ, Zhang Y, Davide JP, Farrell L, Kohl NE, Sepp-Lorenzino L, Lobell RB. Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage. Cancer Cell 2005; 8:49-59.
18. Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell. 2004; 7:637-651.
19. Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science 1989; 246:629-634.
20. Weinert TA, Hartwell LH. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science 1988; 241:317-322.
21. Hoyt MA, Totis L, Roberts BT. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell 1991; 66:507-517.
22. Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell 1991; 66:519-531.
23. Hanks S, Coleman K, Reid S, Plaja A, Firth H, Fitzpatrick D, Kidd A, Mehes K, Nash R, Robin N, Shannon N, Tolmie J, Swansbury J, Irrthum A, Douglas J, Rahman N. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat. Genet. 2004; 36:1159-1161.
24. Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science 1999; 286:971-974.
25. Canman JC, Cameron LA, Maddox PS, Straight A, Tirnauer JS, Mitchison TJ, Fang G, Kapoor TM, Salmon ED. Determining the position of the cell division plane. Nature 2003; 424:1074-1078.
26. Sudo T, Nitta M, Saya H, Ueno HT. Dependence of paclitaxel sensitivity on a functional spindle assembly checkpoint. Cancer Res. 2004; 64:2502-2508.
27. Rieder CL, Schultz A, Cole R, Sluder G. Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J. Cell. Biol. 1994; 127:1301-1310.
28. Li X, Nicklas RB. Mitotic forces control a cell-cycle checkpoint. Nature 1995; 373:630-632.
29. Waters JC, Chen RH, Murray AW, Salmon ED. Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J. Cell. Biol. 1998; 141:1181-1191.
30. Li Y, Benezra R. Identification of a human mitotic checkpoint gene: hsMAD2. Science 1996; 274:246-248.
31. Chen RH, Waters JC, Salmon ED, Murray AW. Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science 1996; 274:242-246.
32. King JM, Nicklas RB. Tension on chromosomes increases the number of kinetochore microtubules but only within limits. J. Cell. Sci. 2000; 21:3815-3823.
33. Biggins S, Murray AW. The budding yeast protein kinase Iβ1/ Aurora allows the absence of tension to activate the spindle checkpoint. Genes Dev. 2001; 15:3118-3129.
34. Meraldi P, Honda R, Nigg EA. Aurora kinases link chromosome segregation and cell division to cancer susceptibility. Curr. Opin. Genet. Dev. 2004; 14:29-36.
35. Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, Su M, Golec JM, Miller KM. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat. Med. 2004; 10:262-267.
36. Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters JM. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell. Biol. 2003; 161:281-294.
37. Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, Taylor SS. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J. Cell. Biol. 2003; 161:267-280.
38. Biggins S, Severin FF, Bhalla N, Sassoon I, Hyman AA, Murray AW. The conserved protein kinase Iβ1 regulates microtubule binding to kinetochores in budding yeast. Genes Dev. 1999; 13:532-544.
39. Cheeseman IM, Anderson S, Jwa M, Green EM, Kang J, Yates JR 3rd, Chan CS, Drubin DG, Barnes G. Phospho-regulation of kinetochore-microtubule attachments by the Aurora kinase Ipl1p. Cell 2002; 111:163-172.
40. Cimini D, Howell B, Maddox P, Khodjakov A, Degrassi F, Salmon ED. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J. Cell. Biol. 2001; 153:517-527.
41. Nicklas RB, Ward SC. Elements of error correction in mitosis: microtubule capture, release, and tension. J. Cell. Biol. 1994; 126:1241-1253.
42. Tanaka TU, Rachidi N, Janke C, Pereira G, Galova M, Schiebel E, Stark MJ, Nasmyth K. Evidence that the Ipl1-Sli15 (Aurora kinase-INCENP) complex promotes chromosome bi-orientation by altering kinetochore-spindle pole connections. Cell 2002; 108:317- 329.
43. Ault JG, Rieder CL, Chromosome mal-orientation and reorientation during mitosis. Cell Motil. Cytoskeleton 1992; 22:155-159.
44. Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat. Rev. Mol. Cell Biol. 2003; 4:842-854.
45. Lampson MA, Renduchitala K, Khodjakov A, Kapoor TM. Correcting improper chromosome-spindle attachments during cell division. Nat. Cell Biol. 2004; 6:232-237.
46. Kapoor TM, Mayer TU, Coughlin ML, Mitchison TJ. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J. Cell. Biol. 2000; 150:975- 988.
47. Peterson JR, Mitchison TJ. Small molecules, big impact: a history of chemical inhibitors and the cytoskeleton. Chem. Biol. 2002; 9:1275-1285.
48. Carter SB. Effects of cytochalasins on mammalian cells. Nature 1967; 213:261-264.
49. Wessells NK, Spooner BS, Ash JF, Bradley MO, Luduena MA, Taylor JT, Wrenn EL, Yamaa, K. Microfilaments in cellular and developmental processes. Science 1971; 171:135-143.
50. Spudich JA, Lin S. Cytochalasin B, its interaction with actin and actomyosin from muscle (cell movement-microfilaments-rabbit striated muscle). Proc. Natl. Acad. Sci. U.S.A. 1972; 69:442-446.
51. Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science 2003; 299:1743-1747.
52. Martineau SN, Andreassen PR, Margolis RL. Delay of HeLa cell cleavage into interphase using dihydrocytochalasin B: retention of a postmitotic spindle and telophase disc correlates with synchronous cleavage recovery. J. Cell. Biol. 1995; 131:191-205.
53. Canman JC, Hoffman DB, Salmon ED. The role of pre- and post-anaphase microtubules in the cytokinesis phase of the cell cycle. Curr. Biol. 2000; 10:611-614.
54. Barr FA, Sillje HH, Nigg EA. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 2004; 5:429-440.
55. Zhang J, Campbell RE, Ting AY, and Tsien RY, Creating new fluorescent probes for cell biology. Nat. Rev. Mol. Cell Biol. 2002; 3:906-918.
56. Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971; 93:2325-2327.
57. Mortlock AA, Jung FH.. Preparation of 4-(N-(5-pyrimidyl)amino) quinolines as inhibitors of aurora 2 kinase, in PCT Int.Appl. 2001, (Astrazeneca AB, Swed.; Astrazeneca UK Limited). Wo. p. 62 pp.
58. Aldridge DC, Armstrong JJ, Speake RN, Turner WB. The cytochalasins, a new class of biologicly active mould metabolites. Chem. Commun. 1967; 1:26-27.