CHEMICAL BIOLOGY
Controlled Drug Delivery: Pharmacokinetic Considerations, Methods and Systems
Zhong Zuo and Vincent H. L. Lee, School of Pharmacy, The Chinese University of Hong Kong, Shatin, N.T., Hong Kong SAR
doi: 10.1002/9780470048672.wecb099
Controlled drug delivery applies interdisciplinary approaches to engineer systems that improve the therapeutic value of drugs. This review addresses the biological basis of drug delivery, the pharmacokinetic/ pharmacodynamic considerations important to the design of controlled release delivery systems, and the methods to fabricate them. The focus of this review will principally be on oral and transdermal applications. Systems at various stages of development for the delivery of more complex molecules, such as proteins, oligonucleotides, genes, and sRNA's will also be discussed.
The frequency of drug dosing is usually determined by the drug’s duration in the body. For drugs that are inherently long lasting, once daily oral dosing is sufficient to sustain adequate drug blood levels and the desired therapeutic effect. Formulation of these drugs as conventional, immediate-release dosage forms is used for the patient. However, many drugs are not inherently long lasting and require multiple doses each day to achieve the desired therapeutic results. Multiple daily dosing often is inconvenient for the patient and can result in missed doses, made-up doses, and patient noncompliance with the therapeutic regimen. In addition, sequential therapeutic blood level peaks and valleys (troughs) are associated with each dose. The traditional approach to modifying drug release is to control the rate of drug delivery, as exemplified by extended-release tablets and capsules, which are commonly taken only once or twice daily. Modern drug delivery systems provide an additional targeted drug release dimension at a specific site in the body, spanning subcellular to organ. Thus, the term “controlled drug delivery” has a much broader meaning than just controlling the rate of drug delivery. Controlled drug delivery to the right site of action at the right time can be achieved by either 1) controlling the release rate and duration or 2) controlling the release site (i.e., localized delivery or targeted diseased organs, tissues or cells). Figure (1) has summarized the scheme for the design of the controlled drug delivery. The commonly used types of controlled drug delivery products in terms of its targeted diseases and specific delivery technology involved are summarized in Table (1).
General Introduction of Pharmacokinetics/Pharmacodynamics
Fate of a drug In vivo: ADME process
After administration, the fate of the drug will be determined by four key steps in vivo, namely, absorption (A), distribution (D), metabolism (M), and excretion (E). Effects of the four steps on the administered drug will influence the levels of drug exposure to the site of action and will eventually influence the pharmacological activities of the drug. The details of the four steps are described briefly as follows.
Absorption (A)
Before a compound can exert its pharmacological effect in tissues, it has to be taken into the bloodstream usually via mucosal surfaces such as the digestive tract for intestinal absorption. In addition, uptake of drugs from the blood stream into target organs or cells needs to be ensured. However, this task is not always easy given the natural barriers that exist, like the blood-brain barrier. Extent of absorption for a drug varies with the way it has been administered. Factors such as poor compound solubility, chemical instability in the stomach, and inability to permeate the intestinal wall can all reduce the extent to which a drug is absorbed after oral administration. Absorption critically determines the compound’s bioavailability. Drugs that absorb poorly when taken orally must be administered in some less desirable way, like intravenously or by inhalation.
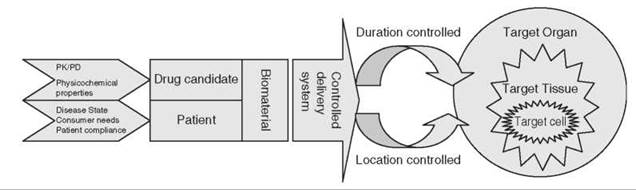
Figure 1. Scheme for the design of controlled drug delivery system.
Distribution (D)
Once the drug gets into the bloodstream, it will be carried to various parts in the body including different tissues and organs, as well as its site of action. Depending on the physicochemical and biological properties of the drug, its degree of distribution to various tissues and organs differs by varying extents.
Metabolism (M)
As soon as the drug enters the body, in addition to being distributed to other parts of the body, it begins to break down. Most small-molecule drug metabolism occurs in the liver by enzymes termed cytochrome P450 enzymes. As metabolism occurs, the parent compound is converted to metabolites, which could be pharmacologically inert or active.
Excretion (E)
Compounds and their metabolites need to be removed from the body via excretion, usually through the kidneys or the feces. Unless excretion is complete, accumulation of foreign substances can affect normal metabolism adversely.
Role of transporters in ADME
The extent of absorption for a drug varies with the way it has been administered. Over the last decade, several important transporters responsible for drug uptake/disposition in various organs have been discovered. They mainly include organic anion transporter, organic cation transporter, multidrug resistant protein, and multidrug resistance-associated protein. Many transporters function in the uptake of drugs into cells, which leads to increased permeability of the drug into cells; other transporters function to export drugs out of cells, which thereby decreases the apparent permeability of the drug. Transport mechanism of drugs and substrate specificities of drug transporters in drug development have become increasingly important. Identification of drugs that are the substrates of the transporters can help to elucidate the pharmacokinetic profiles of these drugs and drug-drug interactions, as well as to improve the therapeutic safety of drugs. Therefore, research that focuses on such membrane transporters is promising, and it leads to rational design strategies for drug targeting delivery. Drug targeting to specific transporters and receptors using carrier-mediated absorption has demonstrated immense significances in the ocular drug delivery (1) and targeted drug delivery across the blood-brain barrier (2).
Concept of pharmacodynamics, biomarkers and pharmacogenetics
Pharmacodynamics is the study of the mechanisms of drug action and the biological and physiological effects of these drugs. Compared with pharmacokinetics, which reveals what the body does to a drug, pharmacodynamics focuses more on what a drug does to the body.
Traditionally, the level of drug in vivo is assumed to be proportional to its pharmacodynamic effect. The identification of various biomarkers provides a more relevant indicator than the in vivo drug concentrations as far as clinical efficacy of a drug is concerned. Biomarkers enable the characterization of patient populations and the quantization of the extent to which new drugs reach intended targets, alter proposed pathophysiological mechanisms, and achieve clinical outcomes. In genomics, the biomarker challenge is to identify unique molecular signatures within complex biological mixtures that can be unambiguously correlated to biological events to validate novel drug targets and to predict drug response. Biomarkers can stratify patient populations or quantify drug benefit in primary prevention or disease-modification studies in poorly served areas such as neurodegeneration and cancer. Appropriateness of biomarkers depends on the strategy and the stage of development, as well as the nature of the medical indication. Biomarkers are perhaps most useful in the early phase of clinical development when measurement of clinical endpoints may be too time-consuming or cumbersome to provide timely proof of concept or dose-ranging information (3).
With the disclosure of the map of the human genome, the effect of human genetic variations on drug metabolism and its related clinical efficacy has become a major concern for clinical practice. Pharmacogenetics is an area of study on the genetic variation in response to drug metabolism with an emphasis on improving drug safety. The aim of drug delivery is to achieve an appropriate drug concentration at the targeted site that can elicit a desired level of response. The concentration of a drug in vivo is highly dependent on the ADME pharmacokinetic processes, whereas the level of response for a drug usually results from the interaction of a drug with a target receptor protein. More and more studies have revealed genetic polymorphisms in drug-metabolizing enzymes, transporters, and target receptors. The genetic basis underlying pharmacokinetic and pharmacodynamic inter-individual variability is essential in the consideration of the design of drug delivery systems (4).
Pharmacokinetic/Pharmaceutical Dosage Form/Pharmacodynamic Considerations in the Design of A Controlled Drug Delivery System
An ideal controlled-release product is expected to release the drug from the dosage form at a predetermined rate, to maintain the released drug at a sufficient concentration in the gastrointestinal fluids, and to provide sufficient gastrointestinal residence time prior to the absorption of the drug. In general, not every drug is suitable to be developed as a controlled-release product.
Traditionally, the pharmacokinetic properties of drug candidates has always played an important role in their design as a controlled release dosage form. From the pharmacokinetic point of view, the best suited drug candidates should have neither very slow nor very fast rates of absorption and excretion. Drugs with slow rates of absorption and excretion with half-lives of greater than 8 hours are usually inherently long acting, and their preparation into extended release dosage forms is not necessary. Drugs with very short half-lives (i.e., < 2 hours) are poor candidates for extended-release dosage forms because of the large quantities of drug required for such a formulation. Another pharmacokinetic consideration is that the drugs prepared in extended-release forms must have good aqueous solubility, must maintain adequate residence time in the gastrointestinal tract, and must be uniformly absorbed from the gastrointestinal tract. In addition, from the classic dosage form design point of view, the drug candidate should be administered in relatively small doses because each oral dosage unit needs to maintain a sustained therapeutic blood level of the drug.
As indicated in Table 1, because of the nature of the duration of action for a controlled drug delivery system, another important pharmacodynamic consideration for drug candidates is that the drugs are used in the treatment of chronic rather than acute conditions. Regardless of how it is achieved, the prolonged and controlled pharmacodynamic effect is the eventual outcome expected for the controlled drug delivery system. The appropriate drug candidate should possess a reasonable therapeutic index (i.e., the median toxic dose divided by the median effective dose). It should be noted that drugs possessing a longer acting quality than indicated by their quantitative pharmacokinetic half-lives might not be good candidates for a controlled drug delivery system either. Therefore, monitoring the controlled release drug more reliably and accurately in vivo is becoming a challenge and the traditional pharmacokinetic/pharmacodynamic correlations as a standard component of drug development are becoming more complicated than what is usually expected (5). An integrated pharmacokinetic and pharmacodynamic approach for rate-controlled drug delivery has been proposed (6). Should pharmacodynamic measurements eventually replace the traditional pharmacokinetic measurements? With the availability of various biomarkers, the measurements of related biomarkers rather than the plasma concentrations of specific drugs are believed to provide more direct and accurate monitoring of the behavior of the controlled drug delivery system in vivo.
Table 1. Summary of representative products using controlled release delivery system
|
|
|
Type of controlled |
|
|
Disease state |
Active ingredient |
delivery system |
Product (manufacturer) |
|
Endocrinology |
|
|
|
|
Postmenstrual syndrome |
Estradiol |
TDD |
Estraderm (Novartis), Vivelle (Novartis), Climara (Berlex) |
|
Hypogonadism in males |
Testosterone |
TDD |
Testoderm (Alza), Androderm (SmithKline Beecham) |
|
Birth control |
Estradiol/Norethindrone |
TDD |
CombiPatch (Noven/Aventis), Ortho-Evra |
|
|
Progesterone |
IUD insert |
Progestersert (Alza) |
|
Hormone replacement therapy |
Estrogen/Progesterone |
TDD |
Nuvelle TS (Ethical Holdings/Schering) |
|
Pain, inflammatory |
|
|
|
|
Moderate/severe pain |
Fentanyl |
TDD |
Duragesic (Janssen) |
|
Central nervous system |
|
|
|
|
Smoking cessation |
Nicotine |
TDD |
Habitrol (Novartis Consumer), Nicoder CQ (SmithKline |
|
|
|
|
Beecham Consumer) |
|
Motion sickness |
Scopolamine |
TDD |
Transderm Scop (Novartis Consumer) |
|
Attention deficit hyperactivity disorder |
Methylphenidate HCL |
Osmotic pump |
Concerta |
|
Cardiovascular disease |
|
|
|
|
Calcium channel antagonist for |
Nifedipine |
Diffusion, osmotic pump, |
Procardia, Procaria XL, Adalat CC |
|
hypertension, ischemic heart |
|
erosion |
|
|
disease, hypertrophic |
|
|
|
|
cariomyopathy, arrhythmias |
|
|
|
|
|
Diltiazem |
Diffusion, erosion |
Cardizem, Cardizem CD, Cardizem SR, Discor XR, |
|
|
Verapamil |
|
Verelan, Calan SR, Isoptin SR |
|
|
Felodipine |
Diffusion, erosion |
Plendil |
|
|
Nicardipine |
Erosion |
Cardene SR |
|
Nitrates for angina pectoris, ischemic |
Nitroglycerin |
TDD (Monolithic and |
Nitrodisc (Searle), Nitro-Dur (Key Pharmaceutical), |
|
heart disease, congestive heart |
|
membrane controlled) |
Transdermal-Nitro (Summit Medical), Deponit (Schwarz |
|
failure. |
|
|
Pharma), Minitran (3 M Pharmaceuticals), etc. |
|
|
Isosorbide mononitrate |
Matrix tablet |
Imdur (Schering-Key) |
|
Hypertension |
Clonidine |
TDD |
Catapres-TTS |
|
Arrhythmia |
Procainamide |
Matrix tablet |
Procan SR (Parke Davis) |
|
|
Disopyramide phosphate |
Multipellet |
Norpace CR |
|
Hypercholesterolemia |
Nicotinic acid |
Polygel matrix delivery system |
Slo-Niacin (Upsher-Smith) |
|
Cancer |
|
|
|
|
Recurrent ovarian cancer |
Doxorubincin |
Stealth liposome |
Doxil, Caelyx |
|
Infectious disease |
|
|
|
|
Fungal infection, AIDS |
Amphotericin B |
Liposome |
AmBisome |
|
Ophthalmic disease |
|
|
|
|
Glaucoma |
Pilocarpine |
Reservoir insert |
Ocusert PILO (Alza) |
Methods and Systems of Controlled Drug Delivery:
Natural and synthetic polymers, both erodible and nonerodible, play an important role in the fabrication of systems to control drug release. The method of combining polymers with a drug candidate, together with the manufacturing process, provides even more control on the release profile desirable of the drug candidate.
Systems to control the release rate and duration of the drug
The rate of drug release from solid dosage forms may be modified by the technologies described below, which in general are based on 1) modifying drug dissolution by controlling access of biologic fluids to the drug through the use of barrier coatings, 2) controlling drug diffusion rates from dosage forms, and 3) chemical reactions or interactions between the drug substance or its pharmaceutical barrier and site-specific biological fluids. The commonly used systems and the related techniques are illustrated as follows.
Coated beads, granules, or microspheres
This system aims to distribute the drug onto beads, pellets, granules, or other particulate systems. The rate at which body fluids can penetrate the coating to dissolve the drug will be adjusted by varying the thickness of the coats and the type of coating material used. The coating techniques include conventional pan-coating or air-suspension coating techniques by which a solution of the drug substance is placed onto small inert nonpareil seeds or beads made of sugar and starch or onto microcrystalline cellulose spheres. Naturally, the thicker the coat, the more resistant to penetration and the more delayed will be the drug release and dissolution. Typically, the coated beads are about 1 mm in diameter. However, the beads are combined to have three or four release groups among the more than 100 beads contained in the dosing unit. This technique provides the different desired sustained or extended release rates as well as the targeting of the coated beads to the desired segments of the gastrointestinal tract. An example of this type of dosage form is the Spansule capsule from SmithKline Beecham (King of Prussia, PA).
Microencapsulated drug
Microencapsulation is a process by which solids, liquids, or even gases may be encapsulated into microscopic sized particles through the formation of thin coatings of “wall” material around the substance being encapsulated. Gelatin is a common wall-forming material, but synthetic polymers, such as polyvinyl alcohol, ethylcellulose, polyvinyl chloride, and other materials, may be used. The typical encapsulation process usually begins with the dissolving of the prospective wall material, such as gelatin, in water. The material to be encapsulated is added and the two-phase mixture is stirred thoroughly. The material to be encapsulated is then broken up into the desired particle size via addition of a solution of a second material, which is usually acacia. This additive material can concentrate the gelatin into tiny liquid droplets. These droplets (the coacervate) then form a film or coat around the particles of the substance to be encapsulated as a consequence of the extremely low interfacial tension of the residual water or solvent in the wall material so that a continuous, tight, film coating remains on the particle. The final dry microcapsules are free-flowing, discrete particles of coated material. Different rates of drug release may be obtained by changing the core to wall ratio, the polymer used for the coating, and the method of microencapsulation. One advantage of microencapsulation is that the administered dose of a drug is subdivided into small units that are spread over a large area of the gastrointestinal tract, which may enhance absorption by diminishing localized drug concentration. An example of a drug commercially available in a microencapsulated, extended-release dosage form is potassium chloride (Micro-K Extencaps; Wyeth, Madison, NJ).
Embedding drug in hydrophilic or biodegradable matrix systems
By this process, the drug substance is combined and made into granules with an excipient material that slowly erodes in body fluids, progressively releasing the drug for absorption. Drugs without excipients provide an immediate drug effect, whereas drug-excipient granules provide extended drug action. The granule mix may be tableted or placed into gelatin capsule shells for oral delivery. Hydroxypropyl methylcellulose (HPMC), which is a free-flowing powder, is commonly used to provide the hydrophilic matrix. Tablets are prepared by distributing HPMC in the formulation thoroughly, preparing the granules by wet granulation or roller compaction, and manufacturing the tablets by compression. After ingestion, the tablet is made wet by gastric fluid, which hydrates the polymer. A gel layer forms around the surface of the tablet and an initial quantity of drug is exposed and released. As water permeates further into the tablet, the thickness of the gel layer is increased, and the soluble drug diffuses through the gel layer. As the outer layer becomes fully hydrated, it erodes from the tablet core. If the drug is insoluble, then it is released as such with the eroding gel layer. Thus, the rate of drug release is controlled by the processes of diffusion and tablet erosion. An example of a proprietary product using a hydrophilic matrix base of HPMC for extended drug release is Oramorph SR Tablets from Roxane Laboratories, Inc. (Columbus, OH), which contains morphine sulfate.
Scientists have been working for decades to expand the number of biodegradable polymers for clinical use, which include polyhydroxyacids, polyanhydrides, polyorthoesters, and polyesteramides. Of these polymers, polylactide (PLA) and poly (D,L-lactide-co-glycolide) (PLGA) are most often used because of their relatively good biocompatibility, easily controlled biodegradability, and good processability. In fact, they belong to one of the two classes of FDA approved synthetic biodegradable polymers to date. These polymers can be used either as matrix devices or as reservoirs. In matrix systems, the drug is dispersed or dissolved in the polymer, whereas the drug is encapsulated in a biodegradable membrane in the reservoir. The release rate of the drug is generally dependent on the degradation of the polymer. Poly(ortho)esters and polyanhydrides erode from their surface, whereas the PLGA and PLA follow bulk erosion (i.e., degradation occurring throughout the whole polymer). By varying the ratios of monomers in the copolymer mix, nearly zero-order drug release produced by matrix degradation at steady rate can be achieved (7).
A few commercial available drug delivery systems are based on biodegradable polymers. Glidadel is a white, dime-sized wafer made up of biodegradable polyanhydride polymer [poly (1,3-bis(carb-oxyphenoxy)propane-co-sebacic acid)] (PPCP-SA) to deliver carmustine for the treatment of brain cancer. ReGel is an aqueous filter sterilizable ABA tri-block polymer system that consists of PLGA and polyethylene glycol (8). Its suitability to provide sustained interleukin-2 (IL-2) delivery for cancer immunotherapy has been reported. Atrigel is a proprietary delivery system that contains PLA or PLGA dissolved in a biocompatible carrier and can be used for both parenteral and site-specific drug delivery. The system is placed in the body using conventional needles and syringes and solidifies on contact with aqueous body fluids to form a solid implant. For a drug incorporated into such polymer solution, it becomes entrapped within the polymer matrix as it solidifies followed by being slowly released as the polymer biodegrades. Products that have already been approved by the FDA using the Atrigel technology include the Eligard (leuprolide acetate for injectable suspension) prostate cancer products that provide systemic release of leuprolide acetate for 1-, 3-, and 4-month duration and the Atridox (8.5% doxycyline) periodontal treatment product for localized subgingival delivery of doxycycline. With the clinical success of the current available products using biodegradable polymers, their additional applications in the delivery of small molecules, peptides, proteins, monoclonal antibodies, and vaccines are anticipated.
Embedding drug in an inert plastic matrix
By this method, the drug is granulated with an inert plastic material, such as polyethylene, polyvinyl acetate, or polymethacrylate, and the granulation is compressed into tablets. The compression of the tablet creates the matrix or plastic form that retains its shape during drug diffusion and its passage through the alimentary tract. An immediate-release portion of the drug may be compressed onto the surface of the tablet. The inert tablet matrix, which is expended of drug, is excreted with the feces. The primary example of a dosage form of this type is the Gradumet from Abbott Pharmaceutical (Abbott Park, IL).
Ion-exchange resins
A solution of a cationic drug may be passed through a column that contains an ion-exchange resin, which forms a complex by the replacement of hydrogen atoms. The resin-drug complex is then washed and may be tableted, encapsulated, or suspended in an aqueous vehicle. The release of the drug is dependent on the pH and the electrolyte concentration in the gastrointestinal tract. Release is greater in the acidity of the stomach than in the less acidic environment of the small intestine. Examples of drug products of this type include hydrocodone polistirex and chlorpheniramine polistirex suspension (Tussionex Pennkinetic Extended Release Suspension; Medeva) and phentermine resin capsules (Ionamin Capsules; Pharmanex, Provo, UT).
Osmotic controlled release systems
The pioneer oral osmotic pump drug delivery system is the Oros system, which was developed by Alza Corporation (Mountain View, CA). The system is composed of a core tablet surrounded by a semi-permeable membrane coating with a 0.4-mm diameter hole in it that was produced by a laser beam. The core tablet has two layers: one contains the drug and the other contains a polymeric osmotic agent. The system operates on the principle of osmotic pressure. When the tablet is swallowed, the semi-permeable membrane permits water to enter from the patient’s stomach into the core tablet, which dissolves or suspends the drug. As pressure increases in the osmotic layer, it forces or pumps the drug solution out of the delivery orifice on the side of the tablet. Only the drug solution can pass through the hole in the tablet. The rate of inflow of water and the function of the tablet depends on the existence of an osmotic gradient between the contents of the bilayer core and the fluid in the gastrointestinal (GI) tract. Drug delivery is essentially constant as long as the osmotic gradient remains constant. The drug release rate may be altered by changing the surface area, the thickness, or the composition of the membrane, and/or by changing the diameter of the drug release orifice; the rate is not affected by gastrointestinal acidity, alkalinity, fed conditions, or GI motility. The biologically inert components of the tablet remain intact during GI transit and are eliminated in the feces as an insoluble shell. This type of osmotic system, which is termed the “gastrointestinal therapeutic system” (GITS; Pfizer, New York), is employed in the manufacture of Glucotrol XL Extended Release Tablets and Procardia XL Extended Release Tablets (Pfizer).
Gastro-retentive systems
Gastric emptying of dosage forms is an extremely variable process, and the ability to prolong and control the emptying time is a valuable asset for dosage forms, which reside in the stomach for a longer period of time than conventional dosage forms. Gastro-retentive systems can remain in the gastric region for several hours, and hence, they significantly prolong the gastric residence time of drugs. Prolonged gastric retention would therefore improve the bioavailability of drugs by reducing drug waste and by improving solubility for drugs that are less soluble in a high pH environment. It has applications also for local drug delivery to the stomach and proximal small intestines. The controlled gastric retention of solid dosage forms may be achieved by the mechanisms of mucoadhesion, flotation, sedimentation, expansion, modified shape systems, or by the simultaneous administration of pharmacological agents that delay gastric emptying. Based on these approaches, classification of floating drug delivery systems has been investigated extensively. Several recent examples have been reported that show the efficiency of such systems for drugs with bioavailability problems. Gastroretention helps to provide better availability of novel products with many therapeutic possibilities and substantial benefits for patients. However, because of the complexity of the pharmacokinetic and pharmacodynamic factors for certain drugs, some researchers suggest that the rationale for continuous administration obtained by controlled-release gastroretentive dosage forms should be assessed and established in vivo (5).
Transdermally controlled drug delivery systems
Because of the large surface area of the skin and its bypass of the liver as a first pass step in metabolism, many drug delivery systems have been developed that control the rate of drug delivery to the skin for subsequent absorption. Effective transdermal drug delivery systems of this type deliver uniform quantities of drug to the skin over a period of time. Technically, transdermal drug delivery systems may be classified into monolithic and membrane-controlled systems (9).
Monolithic Systems
The systems incorporate a drug matrix layer between backing and frontal layers. The drug-matrix layer is composed of a polymeric material in which the drug is dispersed. The polymer matrix controls the rate at which the drug is released for percutaneous absorption. In the preparation of monolithic systems, the drug and the polymer are dissolved or blended together, cast as the matrix, and dried. The gelled matrix may be produced in a sheet or cylindrical form, with individual dosage units cut and assembled between the backing and the frontal layers. Most transdermal drug delivery systems are designed to contain an excess of drug and thus drug-releasing capacity beyond the time frame recommended for replacement.
Membrane-Controlled Transdermal Systems
The system is designed to contain a drug reservoir, usually in liquid or gel form, a rate-controlling membrane, and backing, adhesive, and protecting layers. This system has an advantage over monolithic systems in that as long as the drug solution in the reservoir remains saturated, the release rate of drug through the controlling membrane remains constant. Membrane-controlled systems are generally prepared by preconstructing the delivery unit, filling the drug reservoir, and sealing it off, or by a process of lamination, which involves a continuous process of construction, dosing, and sealing.
The first transdermal therapeutic system designed to control the delivery of a drug to the skin for absorption was developed in 1980 by the Alza Corporation. The system, marketed as Transderm-Scop by CIBA, is a circular, flat adhesive patch designed for the continuous release of scopolamine through a rate-controlling microporous membrane. Soon after, nitroglycerin, which is a drug substance used for the treatment of angina, became another candidate for transdermal delivery. The drug has a relatively low dose, short plasma half-life, high-peak plasma levels, liver first pass metabolism, and inherent side effects when taken sublingually (a popular route for its administration). Since then, several nitroglycerin-containing systems have been developed including: Deponit (Wyeth-Ayerst), Minitran (3 M Riker, St. Paul, MN), TransdermNitro (CIBA-Geigy, Summit, NJ), Nitro-Dur (Key Pharmaceuticals, Kenilworth, NJ), Nitrocine (Schwarz Pharma, Monheim, Germany), and Nitrodisc (Searle). As indicated in Table 1, the application of the transdermal therapeutic system products covers various aspects with the most emphasis on its use in central nervous system disease, cardiovascular disease, and hormonal therapy.
Future developments in the field of transdermal drug delivery should address problems that relate to irritancy, sensitization, and impermeability of the stratum corneum. These hurdles currently exclude several therapeutic entities from delivery via this route. An active area of research is breaching the integrity of the stratum corneum by various means, which include small molecule penetration enhancers, microneedles, and the use of various external energy sources such as electric current and ultrasound (10, 11). The recent approval of lidocaine hydrochloride and epinephrine combined iontophoretic patch for localized pain treatment by FDA has invigorated the gaining interest in iontophoretic drug delivery systems for drug delivery (12). In addition, additional innovations in matrix compound and formulation will likely expand the number of candidate drugs suitable for transdermal delivery (13).
Systems to control the location of drug release: target drug spatially
Liposomes
A liposome is a spherical vesicle with a membrane composed of a phospholipids and cholesterol bilayer. Liposomes can be composed of naturally derived phospholipids with mixed lipid chains, such as egg phosphatidylethanolamine, or of pure surfactants, such as dioleolylphophtidylthanolamine. Liposomes can be made by sonicating phospholipids in water followed by extrusion. Usually, a liposome contains a core of aqueous solution, in which the hydrophilic drug substances could be dissolved. For hydrophobic substances, the drug can be dissolved into the lipid membrane.
Liposomes are used for drug delivery because of their unique properties. To deliver the molecules to sites of action, the lipid bilayer can fuse with other bilayers such as the cell membrane, which delivers the liposomal contents into the cell. For drugs that do not have diffusion capabilities across the cell membrane, formulating the drugs into a liposome would enable them to be delivered past the lipid bilayer. Liposomes can also be designed to have various ways of delivering drugs into cells. It could possess various pH’s, charges, or even made to be within a particular size range such that they would be viable targets for natural macrophage phagocytosis. These liposomes may then be digested while in the magrophage’s phagosome followed by releasing the drug.
Additional advances in liposome research have resulted in the generation of liposomes that can avoid detection by the body’s immune system, specifically the cells of the reticuloendothelial system (RES). These liposomes are known as “stealth liposomes” and are constructed with polyethylene glycol (PEG) studding the outside of the membrane. The PEG coating, which is inert in the body, allows for a longer circulatory life for the drug. In 1995, Doxil, the liposomal doxorubicin, became the first commercially available liposomal anticancer drug. It has an enhanced circulation half-life compared with the free drug because of its surface-grafted polyethylene glycol coating. Doxil passively targets solid tumors, and once the liposomes localize to the tumor interstitial space, the cytotoxic drug is slowly released within the tumor (14). AmBisome is a sterile, nonpyrogenic lyophilized antifungal product for intravenous infusion. It consists of unilamellar bilayer liposomes that are less than 100 nm in diameter with amphotericin B intercalated within the membrane.
Injectable liposome-based drug delivery systems of anticancer drugs are the most widely used drug nanoparticle in cancer (15). In addition to a PEG coating, most stealth liposomes also have some sort of biological species attached as a ligand to the liposome to enable binding via a specific receptor for the targeted drug delivery site. These targeting ligands could be monoclonal antibodies (named as immunoliposome), vitamins, or specific antigens. Targeted liposomes can hone to nearly any cell type in the body and deliver drugs that would otherwise be systemically delivered. It is expected that the side effects of extremely toxic drugs could be drastically reduced if the drugs were only delivered to diseased tissues using such techniques (16).
Nanoparticles
A nanoparticle is a microscopic particle with a diameter less than 100 nm. Nanoparticles were first developed around 1970, and initially they were devised as carriers for vaccines and anticancer drugs. Nanoparticle research is currently an area of intense scientific research because of a wide variety of potential applications in biomedical, optical, and electronic fields. To enhance tumor uptake, the strategy of drug targeting was employed, and as a first important step, research focused on the development of methods to reduce the uptake of the nanoparticles by the RES cells. Simultaneously, the use of nanoparticles for ophthalmic and oral delivery was investigated (17, 18). Recent advancement of nanoparticles and nanosuspensions was caused by their application for pulmonary drug delivery (19, 20).
The size of nanoparticles generally varies from 10 to 1000 nm. The drug is usually dissolved, entrapped, encapsulated, or attached to a nanoparticle matrix, and depending on the method of preparation, nanoparticles, nanospheres, or nanocapsules can be obtained. Nanocapsules are vesicular systems in which the drug is confined to a cavity surrounded by a unique polymer membrane, whereas nanospheres are matrix systems in which the drug is physically and uniformly dispersed. In recent years, biodegradable polymeric nanoparticles using poly(D, L-lactide), PLA, poly(D,L-glycolide) (PLG), PLGA, and poly(cyanoacrylate) (PCA) have attracted considerable attention as potential drug delivery devices in view of their applications in the controlled release of drugs, their ability to target particular organs/tissues, as carriers of DNA in gene therapy, and in their ability to deliver proteins, peptides, and genes through a peroral route of administration. The PLA, PLG, and PLGA polymers have been used as controlled release formulations in parentral and implantation drug delivery applications.
Nanomedicine is undergoing explosive development as it relates to the development of nanoparticles for enabling and improving the targeted delivery of therapeutic and diagnostic agents. The use of nanoparticles for specific delivery to malignant cancers, to the central nervous system, and across the gastrointestinal barriers has received great attention (21). The additional development of nanoparticles is highly dependent on the advancement of the material sciences. For nanotechnology rationally to generate materials useful in human therapy, it will need to progress in full recognition of all the requirements biology places on the acceptability of exogenous materials (22). In particular, nanostructured porous materials, in particular silicon-based photonic and templated materials, offer a degree of control in both the rate and the location of drug delivery that is just beginning to be recognized (23).
Dendrimers
Dendrimers are macromolecules characterized by their highly branched structure and globular shape. They are attractive drug carriers by virtue of the flexibility in precisely controlling their molecular size, shape, branching, length, and surface functionality. An active area of research is the development of nontoxic, biodegradable, and biocompatible dendrimers. Various kinds of dendrimer-drug conjugates have been tried using polyamidoamine, polyphenylether, and polyester dendrimers and anticancer drugs such as doxorubicin, methotrexate, and 5-fluorouracil. The hydrophobic core and hydrophilic shell of dendrimers could encapsulate hydrophobic drugs or diagnostic agents in their interior and release them slowly. With an interior of basic environment, dendrimers could also encapsulate acidic drugs, such as methotrexate.
A unique characteristic of dendrimers is that they can act as a particulate system while retaining the properties of a polymer. Dendrimers can significantly improve pharmacokinetic and pharmacodynamic properties of low molecular weight and protein-based therapeutic agents. The surface modified dendrimers with various kinds of ligands, such as sugars, folate residues, and poly (ethylene glycol), could serve as a drug carrier with targeted cell specificity. Poly (ethylene glycol) modified (PEGylation) dendrimers can generally overcome clearance by RES system. Attachments of targeting moiety on the surface of partially PEGylated dendrimer raised the possibility of a delivery system that can cross biological barriers and deliver the bioactive agent near the vicinity of its target site. Recent successes also demonstrate potential of PEGylated dendrimers in magnetic resonance imaging contrast agent and in carbonyl metallo immunoassay (24, 25).
Erythrocytes/Cells
Erythrocytes, which are also known as red blood cells, have been studied extensively for their potential carrier capabilities for the delivery of drugs and drug-loaded microspheres. Such drug-loaded carrier erythrocytes are prepared simply by collecting blood samples from the organism of interest, separating the erythrocytes from the plasma, entrapping the drug into the erythrocytes, and resealing the resultant cellular carriers. Hence, these carriers are called resealed erythrocytes. The overall process is based on the response of these cells to osmotic conditions. On reinjection, the drug-loaded erythrocytes serve as slowly circulating depots and target the drugs to the RES system. The advantage of erythrocytes as drug carriers is mainly caused by their biocompatibility within the host, particularly when autologous cells are used, and hence triggering an immune response is not possible.
Several methods, that include physical (e.g., electrical-pulse method), osmosis-based systems, and chemical methods (e.g., chemical perturbation of the erythrocyte membrane), can be used to load drugs or other bioactive compounds into erythrocytes.
Resealed erythrocytes have several possible applications in various fields of human and veterinary medicine. Such cells could be used as circulating carriers to disseminate a drug over a prolonged period of time in circulation or in target-specific organs, including the liver, spleen, and lymph nodes. Most drug delivery studies that use drug-loaded erythrocytes are in the preclinical phase. In a few clinical studies, successful results have been obtained.
The ability of resealed erythrocytes to accumulate selectively within RES organs makes them a useful tool for the treatment of hepatic tumors and for the treatment of parasitic diseases. Moreover, the first report of successful clinical trials of the resealed erythrocytes loaded with enzymes for replacement therapy was that of (beta)-glucoserebrosidase in the treatment of Gaucher’s disease, in which the disease is characterized by an inborn deficiency of lysosomal (beta)-glucoserebrosidase in RES cells. This deficiency thereby leads to an accumulation of (beta)-glucoserebrosides in macrophages of the RES. Recently, the same concept was also extended to the delivery of biopharmaceuticals that include therapeutically significant peptides and proteins, nucleic acid-based biologicals, antigens, and vaccines (26). In addition, the possibility of using carrier erythrocytes for selective drug targeting to differentiated macrophages increases the opportunities to treat intracellular pathogens and to develop new drugs. Moreover, the availability of an application that permits the encapsulation of drugs into autologous erythrocytes has made this technology available in many clinical settings and competitive with other drug delivery systems (27).
Summary
During the past four decades, formulations that control the rate and the period of drug delivery (i.e., time-release medications) and that target specific areas of the body for treatment have become increasingly common and complex. Because of researchers’ ever-evolving understanding of the human body and because of the explosion of new and potential treatments that result from discoveries of bioactive molecules and gene therapies, pharmaceutical research hangs on the precipice of yet another great advancement. However, this next leap poses questions and challenges not only for the development of new treatments but also for the mechanisms by which to administer them.
The current methods for drug delivery exhibit specific problems that scientists are attempting to address. Therefore, the goal of all sophisticated drug delivery systems is to deploy medications intact to specifically targeted parts of the body through a medium that can control the therapy’s administration by means of either a physiological or a chemical trigger. To achieve this goal, researchers are turning to advances in the worlds of microtechnology and nanotechnology. During the past decade, systems that contain polymeric materials, such as polymeric microspheres and nanoparticles, have all been shown to be effective in enhancing drug targeting specificity, lowering systemic drug toxicity, improving treatment absorption rates, and providing protection for pharmaceuticals against biochemical degradation.
Peptides, proteins, and nucleotides or DNA fragments are the new generation of drugs. They are becoming attractive because of fast development within the biotechnology field. The administration of such molecules, however, may pose problems such as sensitivity of the molecules to certain temperatures, instability of the molecules at some physiological pH values, a short plasma half-life, and a high molecular dimension, which hinders diffusive transport and makes, at the moment, the parenteral route the only possible way of administering such molecules. Controlled drug delivery that uses the development of new administration routes could be the answer to the problems in administration of these molecules. The rationale of drug delivery is to change the pharmacokinetic and pharmacodynamic properties of drugs by controlling their absorption and distribution. Rate and time of drug release at absorption site could be programmed using a so-called delivery system. Various techniques, that include chemical technologies (such as pro-drugs), biological technologies, polymers, and lipids (such as liposomes) as well as the invention of new routes of administration, have been proposed to achieve controlled drug release. In fact, it could increase drug absorption and reduce the effects of the active ingredient in those regions not requiring therapy. Drug delivery systems that allow for an effective in vivo release of new molecules, such as recombinant anti-idiotypic antibodies with antibiotic activity devoted to the treatment of pulmonary (tuberculosis and pneumocystosis) and mucosal (candidiasis) diseases, fall into that category (28).
References
1. Dey S, Anand BS, Patel J, Mitra AK. Transporters/receptors in the anterior chamber: pathways to explore ocular drug delivery strategies. Expert Opin. Biol. Ther. 2003; 3:23-44.
2. Gaillard PJ, Visser CC, De Boer AG. Targeted delivery across the blood-brain barrier. Expert Opin. Drug Deliv. 2005; 2:299-309.
3. Kuhlmann J. The applications of biomarkers in early clinical drug development to improve decision-making processes. Pharma Center, Bayer Health Care AG, Wuppertal, Germany. Ernst Schering Research Foundation Workshop, Volume 59, 2007. pp. 29-45.
4. Attar M, Lee VHL. Pharmacogenomic considerations in drug delivery. Pharmacogenomics 2003; 4:443-461.
5. Hoffman A, Stepensky D, Lavy E, Eyal S, Klausner E, Friedman M. Pharmacokinetic and pharmacodynamic aspects of gastroretentive dosage forms. Internat. J. Pharmaceut. 2004; 277:141-153.
6. Breimer DD. An integrated pharmacokinetic and pharmacodynamic approach to controlled drug delivery. J. Drug Target. 1996; 3:411-415.
7. Kwon GS, Furgeson DY. Biodegradable polymers for drug delivery systems. Biomed. Polym. 2007; 83-110.
8. Kissel T, Li YX, Unger F. ABA-triblock copolymers from biodegradable polyester A-blocks and hydrophilic poly (ethylene oxide) B-blocks as a candidate for in situ forming hydrogel delivery systems for proteins. Adv. Drug Deliv. Rev. 2002; 54:99-134.
9. Moore L, Chien WY. Transdermal drug delivery: a review of pharmaceutics, pharmacokinetics, and pharmacodynamics. Crit. Rev. Ther. Drug Carrier Sys. 1988; 4:285-349.
10. Gill HS, Prausnitz MR. Coated microneedles for transdermal delivery. J. Control. Rel. 2007; 117:227-237.
11. McKinnie J. Transdermal market: the future of transdermal drug delivery relies on active patch technology. Drug Deliv. Tech. 2006; 6:54-58.
12. Dixit N, Bali V, Baboota S, Ahuja A, Ali J. Iontophoresis - an approach for controlled drug delivery: a review. Curr. Drug Deliv. 2007; 4:1-10.
13. Hadgraft J, Lane ME. Passive transdermal drug delivery systems: recent considerations and advances. Am. J. Drug Deliv. 2006; 4:153-160.
14. Allen TM, Cheng WWK, Hare JI, Laginha KM. Pharmacokinetics and pharmacodynamics of lipidic nano-particles in cancer. Anti-Cancer Agents Med. Chem. 2006; 6:513-523.
15. Gabizon AA, Shmeeda H, Zalipsky S. Pros and cons of the liposome platform in cancer drug targeting. J. Liposome Res. 2006; 16:175-183.
16. Gabizon AA. Applications of liposomal drug delivery systems to cancer therapy. Nanotech. Cancer Ther. 2007; 595-611.
17. Zimmer A, Kreuter J. Microspheres and nanoparticles used in ocular delivery systems. Adv. Drug Deliv. Rev. 1995; 16:61-73.
18. Vandervoort J, Ludwig A. Ocular drug delivery: nanomedicine applications. Nanomedicine 2007: (1):11-21.
19. Ozeki T, Okada H. Nanoparticle-containing microspheres for pulmonary drug delivery. Yakuzaigaku 2006; 66:249-253.
20. Chow AHL, Tong HHY, Chattopadhyay P, Shekunov BY. Particle Engineering for Pulmonary Drug Delivery. Pharm. Res. 2007; 24:411-437.
21. Emerich DF, Thanos CG. Targeted nanoparticle-based drug delivery and diagnosis. J. Drug Target. 2007; 15:163-183.
22. Petrak K. Nanotechnology and site-targeted drug delivery. J. Biomat. Sci. 2006; 17:1209-1219.
23. Cunin F, Li Y, Sailor MJ. Nanodesigned pore-containing systems for biosensing and controlled drug release. Bio. Biomed. Nanotech. 2006; 3:213-222.
24. Bai SH, Thomas C, Rawat A, Ahsan F. Recent progress in dendrimer-based nanocarriers. Crit. Rev. Ther. Drug Carrier Sys. 2006; 23:437-495.
25. Patri AK, Kukowska-Latallo JF, Baker Jr. Targeted drug delivery with dendrimers: comparison of the release kinetics of covalently conjugated drug and non-covalent drug inclusion complex. Adv. Drug Delivery Rev. 2005; 57:2203-2214.
26. Hamidi M, Zarrin A, Foroozesh M, Mohammadi-Samani S. Applications of carrier erythrocytes in delivery of biopharmaceuticals. J. Control. Rel. 2007; 118:145-160.
27. Rossi L, Serafim S, Pierige F, Antonelli A, Cerasi A, Fraternale A, Chiarantini L, Magnani M. Erythrocyte-based drug delivery. Exp. Opin. Drug Deliv. 2005; 2:311-322.
28. Conti S, Polonelli L, Frazzi R, Artusi M, Bettini R, Cocconi D, Colombo P. Controlled delivery of biotechnological products. Curr. Pharm. Biotech. 2000; 1:313-323.
Further Reading
Katz B, Rosenberg A, Frishman WH. Controlled-release drug delivery systems in cardiovascular medicine. Am. Heart J. 1995; 129:359-368.
Mayer PR. Pharmacodynamic and pharmacokinetic considerations in controlled drug delivery. Control. Drug Deliv. 1997; 589-595.
Suzuki H, Nakai D, Seita T, Sugiyama Y. Design of a drug delivery system for targeting based on pharmacokinetic consideration. Adv. Drug Deliv. Rev. 1996; 19:335-357.
See Also
Drug Delivery
Controlled Drug Delivery: Pharmacokinetic Considerations, Methods and Systems