CHEMICAL BIOLOGY
Coupling Methods for Peptide Synthesis
N. Leo Benoiton, University of Ottawa, Ottawa, Ontario, Canada
doi: 10.1002/9780470048672.wecb101
Peptide-bond formation requires activation of a carboxyl group followed by aminolysis of the activated carbonyl. The reagents and methods employed for activation, the intermediates and activated forms generated and their aminolysis are described. Rationalization of the strategy employed to avoid isomerization during coupling is presented. Approaches for minimizing aggregation that interferes with coupling are also given.
Peptides are employed for therapeutic purposes and for research in the development of new drugs, vaccines, biomarkers, diagnostic agents, and so on. The demand for these peptides has experienced double-digit annual growth over recent years (1). Most peptides are obtained by chemical synthesis. Chain assembly is effected by successive addition of single residues always starting from the carboxy-terminus, which eliminates the danger that isomerization might occur during coupling. If the peptide is long enough, then two or more segments may be combined to produce the final product. Moreover, the chain may be assembled in solution or on a solid phase. Many methods are available for linking two amino acids together. Each has its own peculiarities as well as favorable and unattractive features. Some methods of coupling are of general use, some are not applicable to solid-phase synthesis, and some are employed only for specific purposes. The choice of method depends on circumstances that vary considerably; every method seems to find its place among the armaments employed for synthesis. An intriguing observation is that five different coupling methods are involved in the synthesis in kilogram amounts of the nine-residue drug Atisoban (2).
Peptide-Bond Formation
A peptide bond is formed by reaction of the carboxyl group of one amino acid with the amino group of a second amino acid (Fig. 1A). A combination of the two occurs when the carboxyl group is activated, which means it has been rendered deficient in electrons or electrophilic, and the amino group is electron rich or nucleophilic. The amino group is nucleophilic when it is not protonated; the carboxyl group is activated by affixing an electron-withdrawing moiety to it. The latter is achieved by use of compounds called coupling reagents or by one or more chemical reactions. Peptide-bond formation is slower when the activated residue is valine, isoleucine, or threonine protected on the side chain; these are known as hindered residues. During the process of aminolysis, a competing reaction may occur (Fig. 1B) because the carbonyl on the amino group of the activated residue is also nucleophilic. The result is the formation of a five-membered cyclic structure, which is referred to as a 5(4H)-oxazolone that contains nitrogen and oxygen atoms and a carbonyl group, with substituents on carbons 2 and 4. The substituent at C-4 is the side chain of the amino acid; the substituent at C-2 originates from the acyl group of the activated residue. When the acyl group is another amino-acid residue, that is, if the activated component is a peptide, then the structure is a 2-alkyl-5(4H)-oxazolone that is chirally unstable. It readily tautomerizes (Fig. 1F) to give a mixture of the two enantiomers of the oxazolone. When the acyl group is that of the protector of an amino acid, the ox- azolone that is formed is a 2-alkoxy-5(4H)-oxazolone, which is chirally stable and does not isomerize (3). These alkoxycarbonyl protectors (ROCO, often referred to as “urethane” protectors, which is incorrect; urethane = ROCONH-), namely benzyloxycarbonyl (Cbz or Z; benzyl = C6H5CH2) cleaved by hydrogenolysis, tert-butoxycarbonyl (Boc) cleaved by acidolysis, and 9-fluorenylmethoxycarbonyl (Fmoc) cleaved by beta-elimination have been designed, in that order, so that the activated residue does not enantiomerize during couplings. The effect of the tautomerization of the 2-alkyl-5(4H)-oxazolone is the production (Fig. 1E') of some other isomer of the peptide product because the oxazolone also undergoes aminolysis. Hence, coupling of an N-alkoxycarbonylamino acid occurs without enantiomerization of the residue because even if the 2-alkoxy-5(4H)-oxazolone is formed, it does not isomerize. However, there is a danger to produce epimeric peptides during the coupling of an N -protected peptide because of the severe tendency of the activated peptide to form the 2-alkyl-5(4H)-oxazolone that isomerizes readily and is also a precursor of the product. In all cases, the product of isomerization is a mixture of epimeric peptides. When the activated residue is isoleucine or threonine that have two stereogenic centers and when the activated component is a peptide, which does not have glycine at the terminus, the process is an epimeriza- tion. When the activated component bears an N -alkoxycarbonyl group and the amino acid is other than glycine, isoleucine, or threonine, the process is an enantiomerization (4), but it is also referred to as racemization. The process of isomerization of an activated peptide is also often referred to as racemization, but it is careless usage of the term and an unwarranted extension of the meaning of the word.
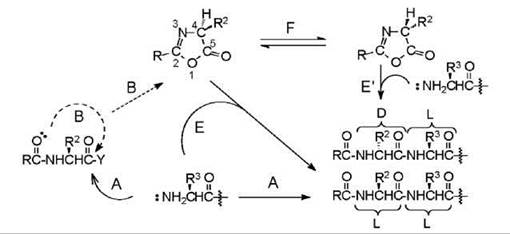
Figure 1. Coupling in peptide synthesis. Aminolysis (A) of the activated residue gives the L-L peptide. A competing intramolecular reaction (B) may occur giving the 5(4H)-oxazolone. When R = alkoxy (BzlO, tBuO, or FmO), the oxazolone is aminolyzed (E) giving the L-L peptide. When R = peptidyl (-Xaa-NHCHR3) the oxazolone tautomerizes (F) and is then aminolyzed (E, E’) giving also the D-L peptide.
Activated Forms
A multitude of activated forms of amino acids and peptides is available (Fig. 2), which have varying stabilities. Some are shelf-stable reagents; some can be prepared and manipulated, but they are not stable enough to be stored; and others are intermediates that have been postulated to explain the results. In fact, the most popular coupling reagents generate activated forms that have never been detected; hence, they are hypothesized structures. Noteworthy among these structures is the O-acylisourea that comes from carbodiimides (5), the acyloxyphosphonium cation that comes from the phosphonium-salt reagents, and the O-acyluronium cation that comes from the uronium-salt reagents (Fig. 2). The mixed (MxAn) and symmetrical (SyAn) anhydrides are compounds that can be purified and characterized, but they are usually prepared and used immediately. The SyAn is the anhydride formed from the combination of two molecules of N-alkoxycarbonylamino acid. The MxAn is formed by reaction of the latter with an alkyl chloroformate (chlorocarbonate). Both structures occasionally surface as intermediates in reagent-mediated reactions, namely the SyAn in carbodiimide-mediated reactions and the MxAn in EEDQ-mediated (see Fig. 4) reactions. Acyl azides and halides are prepared in solution and used immediately. Some activated esters are shelf-stable reagents. They are of two kinds, which originate from substituted phenols and substituted hydroxamic acids (Fig. 3).

Figure 2. Activated forms of N-alkoxycarbonylamino acids (RCO = R1OCONHCHR2CO) and Na-protected peptides. Some are aminolyzed (A) to give the peptide, and some may generate (B), a second activated form that is also a source of the product. Experience indicates that acyl halides and symmetrical anhydrides of peptides do not exist.
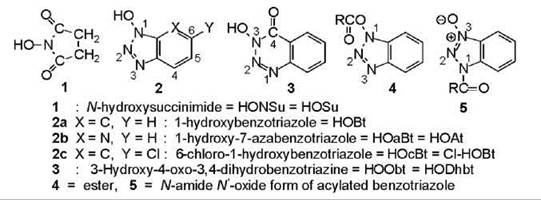
Figure 3. Auxiliary nucleophiles (additives) that suppress side reactions by reacting with activated intermediates, and the two acylated forms of benzotriazole that are produced.
Auxiliary Nucleophiles
Peptide-bond formation involves reaction of the nucleophilic amino group of one residue with the activated carboxyl group of a second residue (Fig. 1). In many cases, the activated residue reacts first with another molecule to produce a compound that is activated by another moiety; the latter then undergoes aminolysis to give the peptide. These molecules are referred to as auxiliary nucleophiles. The original ones were incorporated into coupling reactions when it was realized that their presence had beneficial effects. The common auxiliary nucleophiles (Fig. 3) (6-9), which are also known as additives, appear in Fig. 4. Each possesses a hydroxyl group; all except the HOSu are slightly acidic with p Ks in the 3.4-4.6 range. They participate in coupling reactions in one of two forms. Either they are added in their original form HOW, or they are present by virtue of the fact that the coupling reagent employed is composed of the deprotonated moiety -OW attached to a positively charged atom that is a strong electrophile (Fig. 5). HOSu serves primarily as a nucleophile for the preparation of activated esters (10); that is, it is added to an activated amino acid without the simultaneous presence or subsequent addition of an amino-bearing residue. The other auxiliary nucleophiles are added to carbodiimide-mediated reactions to suppress the side reactions of isomerization (7) (Fig. 1) and N-acylurea formation (Fig. 4). They protonate the intermediates that are formed, namely the O-acylisourea (Fig. 4) and the oxazolone (Fig. 1), which prevents their rearrangement and tautomerization, respectfully, that lead to undesired consequences. The protonated intermediates are strong nucleophiles that readily yield the peptide product. Hence, the additives trap activated intermediates that prevent the side reactions. When the additives are incorporated into coupling reagents, they are released as the anion - OW as soon as the reaction is initiated. The anion is a strong nucleophile that can react with or trap any activated intermediates that are formed. The compounds generated are activated esters that undergo aminolysis to give the peptide. The traditional additive has been HOBt (7); HOAt (HOaBt) (9) is of more recent development. HOObt (8) was not employed in the past because of the known side reaction of aminolysis at the carbonyl of the additive. However, this reaction occurs in negligible amount, and the additive has received renewed interest. Although it was dormant for many years, HOcBt (7) has surfaced with a vengeance as it is being employed in multikilogram amounts in the synthesis of cephalosporin and the fusion inhibitor Fuzeon. It and HOAt are often superior to HOBt as additives; their hydroxyl groups are slightly more acidic, which provides better leaving groups in the esters. HOcBt is less expensive. The addition of HOBt to a carbodiimide-mediated reaction minimizes the dehydration of side-chain amides that occurs in its absence.
Coupling Methods
Peptide-bond formation is achieved in two ways. In the first approach, a reagent is added to a mixture that contains a component with a free carboxyl group and one with a free amino group. Such coupling reagents include carbodiimides, EEDQ (Fig. 4), and onium-salt based reagents (Fig. 5). The other approach involves preparing the activated component after which the amino-bearing component is added. Here, one can distinguish two separate cases. In one case, the activated form is a shelf-stable compound such as an activated ester or an N-carboxyanhydride (NCA). In the other case, the activated form is employed as soon as it is prepared. Examples include MxAn as well as SyAn and azides.
Dialkylcarbodiimides
The compounds implicated in carbodiimide-mediated reactions (11, 12) appear in Fig. 4A. The reaction is postulated to be initiated by protonation of one of the slightly basic nitrogen atoms by the acid followed by attack at the central carbon atom by the acid anion that generates the O-acylisourea (OAIU). Aminolysis the OAIU gives the peptide with liberation of N,N'-dialkylurea. A side reaction of N-acyl-N,N'-dialkylurea (NAU) formation by O-to-N acyl shift of the OAIU can occur. The latter is avoided by adding an auxiliary nucleophile that protonates intermediates. The acyl shift is promoted by heat and tertiary amine that assures a deprotonated nitrogen atom, and delay in the aminolysis caused by bulkiness in the reacting residues. The three common carbodiimides are dicyclohexyl-carbodimide (R4 = R5 = C6Hn, DCC) (11) giving urea DCU, diisopropylcarbodimide (R4 = R5 = C(CH3)2, DIC) giving urea DIU, and ethyl(dimethylaminopropyl)carbodiimide hydrochloride (R4 = C2H5, R5= (HCl. (CH3)2NCH2CH2CH2, EDC) giving urea EDU (12). All carbodiimides are allergenic, which cause skin irritation, so they should be handled with care. DCC is an inexpensive, brittle solid that produces DCU that is bulky and insoluble in most solvents. DIC is a moderately expensive liquid employed in solid-phase synthesis because DIU is soluble in organic solvents. Contact of eyes with fumes from spills of DIC can cause temporary blindness. The soluble EDC is excellent for synthesis in solution because EDU and the NAU are soluble in water. It is available as crystals that are expensive. Reactions are usually effected by adding the reagent to a mixture of the reacting components and the auxiliary nucleophile if desired. In a variant, the acid is allowed to react with itself and possibly an additive after which the amino-containing component is added. In this case, the N-alkoxycarbonylamino acid is converted into the SyAn and the activated ester, respectively, prior to the aminolysis. The efficiency of a carbodiimide-mediated coupling of a peptide that is effected in the presence of an auxiliary nucleophile is enhanced by the presence of cupric ion (13). Cupric ion prevents a 2,4-dialkyl-5(4H)-oxazolone from isomerizing (13).
1-Ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ)
This reagent (14) (Fig. 3B) is employed in the same way as carbodiimides and occasionally by operators who have developed a skin irritation after contact with DCC. The acid displaces the ethoxy group after which spontaneous rearrangement to the MxAn occurs with expulsion of quinoline. Aminolysis of the MxAn gives the peptide. Unavoidable aminolysis at the other carbonyl gives a small amount of irreversibly substituted amino component as impurity.
Symmetrical anhydrides (Fig. 2)
In a variant (15) of the carbodiimide method, two moles of an N-alkoxycarbonylamino acid are reacted with one mole of DCC in dichloromethane (DCM). After 30 minutes, the DCU is removed by filtration and the solution is mixed with the aminolyzing component. The DCM is sometimes replaced with dimethylformamide (DMF) for the aminolysis step. SyAn formation is very slow in DMF. The method is wasteful of substrate but gives clean reactions. Some SyAn have been isolated and stored (16, 17), but the practice is no longer in vogue. SyAn are less reactive and hence more selective than OAIU; they do not acylate hydroxy groups that have not been deprotonated. They are especially effective for acylating secondary amines such as N-methylamino-acid residues.
Mixed anhydrides (Fig. 2)
The MxAn method is popular for synthesis in solution; it is inexpensive and simple to effect. Chloroformate R6OCOCl [R6 = ethyl, C2H5 or isobutyl, CH(CH3)2] is added to a small excess of the acid at —5 °C in the presence of tertiary amine and after 30-60 seconds, the aminolyzing component is added (18). The reaction is complete within 30-60 minutes. Traditional solvents have been tetrahydrofuran and DMF; the favored tertiary amine is N-methylmorpholine (NMM). NMM is believed to act as an acceptor of the acyl group that forms the acylmorpholinium ion (RCO-N+MM), which is the acylating reagent. It was thought for many years that halo-containing solvents were unsuitable; however, DCM is a good solvent provided the tertiary amine is not triethylamine as this combination retards the formation of the MxAn (19). Strictly anhydrous conditions are also not essential because a MxAn can be purified by washing it with aqueous solutions (19). MxAn have not been employed in solid-phase synthesis because a side reaction of aminlyosis occurs at the wrong carbonyl (Fig. 4B) in the range of 1-10% that is unavoidable. The larger amounts develop when the activated residue is hindered. The urethane produced is stable to all conditions used for deprotection; hence, the impurity cannot be readily eliminated. Isopropyl chloroformate seems to be a superior reagent for MxAn reactions (20). MxAn can be employed for acylation of an amino acid or peptide with an unprotected carboxyl group in polar or partially aqueous solvent
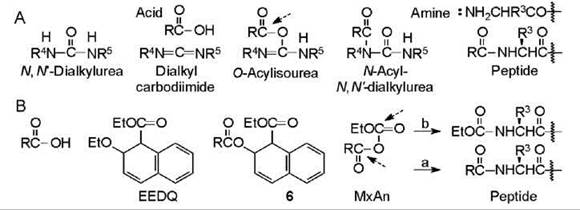
Figure 4. (A) Compounds implicated in carbodiimide-mediated reactions. The acid is postulated to add to the carbodiimide generating the O-acylisourea. Aminolysis occurs at the point indicated by the dashed arrow. (B) Compounds implicated in EEDQ-mediated reactions. The acid is postulated to displace the ethoxy of the reagent generating 6 which rearranges to the MxAn. Aminolysis (a) gives the desired peptide and (b) the ethoxycarbonylated nucleophile as impurity.
Phosphonium and uronium salt-based reagents
A revolution in coupling occurred with the introduction of BOP (21) (Fig. 5). Numerous related reagents followed (Fig 5) (22, 23). All reagents react according to the same mechanism. An equi-mixture of the reagent, acid, and amino-containing component is prepared, and two equivalents of a tertiary amine are added. No reaction takes place without the tertiary amine, which creates the carboxylate ion RCO2—. The anion displaces the ring structure of the reagent generating the acyloxy-phosphonium (Fig. 1) or -carbenium cation, never detected but postulated, which then undergoes nucleophilic attack by one or both of the available species, the amino-containing component that produces peptide and the benzotriazolyl BtO- or equivalent anion AtO- that gives the activated ester RCO2Bt, RCO2At, and so on, which undergoes aminolysis to produce peptide. The HOBt/HOAt that is liberated is neutralized by the second equivalent of base that had been added. Many assume that the activated ester is the source of the peptide, but indisputable evidence suggests peptide originates also from some other intermediate (24), which means the initial intermediate. The preferred tertiary amine is diisopropylethylamine (DIEA) and not NMM as originally employed because the more basic amine is necessary to drive the reaction to completion. A variant involves the addition of an equivalent of HOBt or HOAt to encourage formation of the activated ester. However, this ester formation is not always beneficial as the benzotriazolyl esters of hindered residues undergo aminolysis much more slowly to the point that the benzotriazolyl ester of N-protected-N-methylvaline is resistant to aminolysis (25). The hexamethyl-substituted reagents generate the toxic hexamethylphosphor-trisamide as side product. The tripyrrolidino-containing reagents generate an amide that is environmentally acceptable. The tetramethylurea that is produced from the uronium-based reagents is soluble in water and easy to dispose of. The phosphonium-based reagents are more reactive than the carbenium-based reagents, hence, they are less stable for storage. For this reason, the carbenium (uronium)-based reagents are more attractive for solid-phase synthesis. The chemistry of the uronium-based reagents is complex. In fact, the BTU of HBTU and TBTU does not have the structure given in Fig. 5, but it is 1-(bis(dimethylamino)methylene-1-H-benzotri azolylinium 3-oxide, that is, the tetramethycarbenium moiety is linked to the other nitrogen atom of the ring, analogous to structure 5 (Fig 3) (26). Acylated HOBt had originally been shown to exist in solution as two forms in equilibrium, the ester (RCO2-Bt) and the N-acyl N'-oxide (RCO-Bt→O) (6). In model experiments, the uronium forms of HBTU and HATU reacted more quickly and led to less epimerization than the guanidinium forms (27). Other reagents are continuously being developed, typical examples being BtO-CH=N+ Me2 . PF6- (28) and PyCOP (Fig. 5) (29). Reagents based on HODhbt Fig 3 have also been described. All have their unique properties. The azabenzotriazolyl-containing reagents seem to be particularly effective for performing difficult couplings (30).
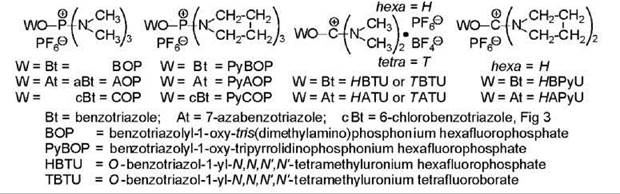
Figure 5. Structures and examples of nomenclature for phosphonium and carbonium salt-based reagents. Rules of nomenclature dictate that the latter be named as modified ureas; see text for revised structures; ''ino'' indicates a ring linked at the nitrogen atom.
Acyl halides (Fig. 2)
A straightforward method of coupling involves conversion of an Fmoc-amino acid (Fmoc-Xaa-OH) to the chloride Fmoc-Xaa-Cl, using thionyl chloride (SO2Cl) in DCM (31, 32) or triphosgene (O=C(OCCl3)2, which is a source of phosgene (COCl2) (33). Only the latter allows preparation of derivatives with acid-sensitive side-chain protectors tBu but not trityl. Also available are Cbz-, Boc- and Fmoc-amino-acid fluorides obtained from the acids using cyanuric fluoride (cyclic C3N3F3), diethylaminosulfur trifluoride (DAST, Et2NSF3), or tetram- ethylfluoroformamidium hexafluorophosphate (TFFH, Me4 N2C+F. HF6- which is activated in DMF by tertiary amine. The amino-containing component is added after removal of excess reagent and side products. TFFH is employed for generating the fluoride in the presence of the incoming nucleophile. The Fmoc-Xaa-Cl are highly activated reagents that generate the strong acid HCl on aminolysis. Neutralization by tertiary amines causes formation of the 5(4H)-oxazolone that is aminolyzed more slowly, so it is best to avoid this structure by use of KOBt for neutralization or by destruction (reduction) of the acid with powdered zinc (34). For this and other reasons, Fmoc-Xaa-Cl are not used routinely, but they are particularly suited for couplings between hindered residues, and for esterifying a hydroxymethyl-linker-resin. Acyl fluorides are less reactive, hence, are more stable to oxygen nucleophiles such as water or methanol; they have a lower tendency to cyclize to the 5(4H)-oxazolone and can be employed in the absence of base. Their properties resemble those of activated esters. They are also suited for coupling hindered residues, but the possible deleterious effects of prolonged exposure to base because of their lesser reactivity must not be disregarded.
Acyl azides (Fig. 2)
The preparaton of acyl azides involves conversion of acid RCO2H to ester RCO2Me and subsequent hydrazinolysis with excess N2H4 to give acyl hydrazide RCONHNH2 (35). The latter is purified by crystallization and treated at +5 °C with a solution of aqueous AcOH that contains NaNO2 or tert-butylnitrite. The acyl azide (RCON3) that is formed is extracted into an organic solvent, which is dried and added to the aminolyzing component. An acyl azide can be produced directly from the anion of an acid in the presence of the aminolyzing component by use of diphenyl phosphorazidate [(PhO)2PON3] (36), which is a reagent that is especially good for converting a linear peptide to a cyclic peptide. The acyl azide reaction is generally applicable to Boc- or Z-Xaa-OH, which includes instances when Xaa = Ser, Thr, or His with unprotected side chains. For coupling peptides, activation of the hydrazide is performed with tert-butyl nitrite (tBuNO2) and nitrosyl chloride (NO2Cl) (37). Because hydrazine destroys protectors such as trifluoroacetyl (CF3C=O), o-nitrophenysulfanyl (O2NPhS), and nitro of nitroarginine, a variant that circumvents the problem assembles the chain starting with a protected hydrazide (H-Xaa-NHNH-O2CR2) obtained in solution or on a solid support (H-Xaa-NHNH-linker-resin) (38). Aminolysis of an acyl azide is very slow. tert-Butyl nitrite with HOAt converts the hydrazide to an activated ester that allows aminolysis to occur in 4 hours instead of 2-3 days (39). Acyl azides are sensitive to tertiary amines; however, they do not form oxazolones, hence, proper manipulation of the coupling species just about guarantees products that are enantiomerically pure. Caution: Azides tend to explode under certain conditions.
Activated esters (Fig. 2)
Two types of activated esters exist, those in which the activating moiety is a substituted phenol and those in which the activating moiety is a substituted hydroxylamine (Fig. 2). Common examples of the former are p-nitrophenyl, pentachlorophenyl and pentafluorophenyl; common examples of the latter are succinimido, benzotriazolyl, 7-azabenzotriazolyl, and 4-oxo-3,4-dihydrobenzotriazinyl (Fig. 3). All (RCO2W) react with an amino group (NH2R') to produce tetrahedral intermediate R(-O)C(OW)-N+H2R' that collapses to the product RC(=O)NHR' at a speed that is the limiting rate of the reaction (40). The reaction is catalyzed by mild acid, and the rate is much greater in polar solvents; a difference of a hundred times for reactions in DMF versus DCM is typical. Penta-substituted phenyl esters are more reactive than p-nitrophenyl esters. With the exception of succinimido esters, the aza-containing esters are encountered as intermediates in reactions mediated by carbodiimides or onium-salt reagents (see above). Occasionally, the intermediate is generated intentionally, by adding the amino-containing component after a delay. These esters are highly reactive species because of the phenomenon of an- chimeric assistance, which is a favorable juxtaposition of the potentially reactive atoms (41). Succinimido esters (10) are unique in that the moiety liberated, HONSu, is soluble in water; hence, they are easier to purify. They are less reactive and are more stable than other aza-containing esters. Activated esters are usually prepared by reaction of the acid with the phenol or HONSu and a carbodiimide, or by ester interchange. Caution must be taken in the preparation of succinimido esters. Use of an excess of HONSu to drive the reaction can result in the formation of an impurity that develops from the condensation of three moles of HONSu, SuNO2C-NHCH2CH2C(=O)-ONSu (42), which is an activated ester that causes the incorporation of beta-alanine into a chain. p-Nitrophenyl esters are employed instead of DCC for activating asparagine and glutamine derivatives during chain assembly to avoid the dehydration of the carboxamide groups by the carbodiimide. Pentafluorophenyl esters are employed for temporary protection during the formation of a bond between the anomeric carbon of a hexose and the nitrogen on the side chain of asparagine. Succinimido esters are useful for acylating amino-acid or peptide anions in polar or partially aqueous solvents. 2-Chloro-4,6-dimethoxy-1,3,5-triazine is a reagent that produces superactive triazine esters from acids (43).
Amino-acid N-carboxyanhydrides (Fig. 2)
A unique form of activation of an amino acid but not of a peptide is the N-carboxyanhydride (NCA) (Fig. 2), which is the cyclic anhydride formed between the carboxyl group and a carboxyl group bound to the amino group. These anhydro N-carboxyamino acids are obtained by reacting the amino acid with phosgene in tetrahydrofuran (44). Addition of Xbb-NCA to H-Xaa- gives peptide H-Xbb-Xaa- after acidifiction to release CO2 from the amino-terminus (45). It is possible that the NCA oligomerizes, so the reaction must be effected under very controlled conditions of pH and temperature. For this reason, NCAs are not employed routinely. An option that eliminates the dimerization is use of the N -alkoxycarbonyl- or “urethane-” protected amino-acid NCA, Boc, Cbz or Fmoc serving as useful substituents (46). These structures also are not employed routinely but have proved advantageous in certain cases. They decompose in the presence of tertiary amine.
Tactics for Avoiding Aggregation
The coupling of a protected residue to a peptide chain is inefficient or occasionally fails once the chain has been extended to five or more residues (47). Based on the observation that replacement of the pertinent residue by a secondary amino acid eliminates the difficulty (48), it has been postulated that the problem occurs because of the chain folding on itself or the binding of two chains because of the tendency of functional groups to form hydrogen bonds (>C=O - - - NH<). The resulting aggregation interferes with the merger of the reacting groups. Tactics to prevent the formation of hydrogen bonds and hence aggregation have been developed. These tactics involve elimination of the hydrogen atom on a peptide bond at the fifth or sixth residue of a chain and the use of microwave energy. Peptides that contain serine and are insoluble in aqueous medium can be administered to an organism in the soluble O-acyl form H-Ser(peptide1)-peptide2-OH that immediately undergoes O-to-N acyl shift to H-peptide1-Ser-peptide2-OH at above neutral pH (49).
N-Alkylation of the Peptide Bond
Elimination of the hydrogen atom at a peptide residue to prevent hydrogen-bond formation involves replacing it with a functional group that can be removed at the end of the synthesis. The protector must be stable to all the conditions employed during assembly of the chain, be easy enough to introduce on to the amino group, and must allow efficient coupling of the residue bearing between the two N -protectors as well as the subsequent aminolysis reaction by the N-alkylamino-acid derivative. Producing such a protector has not been an easy task. The protector of choice for Fmoc/tBu chemistry is 2-hydroxy-4-methoxybenzyl (Hmb) (50) (see Fig. 6). Coupling is achieved by HOBt-assisted aminolysis by H-Xaa- of the pentafluorophenyl ester of the N,O-disubstituted-derivative 8 giving 9. The two protectors are removed, the next residue is introduced as the SyAn [(Fmoc-Xcc)2O], which acylates the 2-hydroxy group of the Hmb giving 10. The residue then migrates spontaneously to the amino group via the O-to-N acyl shift giving N -substituted peptide 11. The protector of choice for Boc/Bzl chemistry is 2-hydroxybenzyl.

Figure 6. Synthesis of a peptide chain with a bond protected by the 2-hydroxy-4-methoxybenzyl (Hmb) group.
Pseudo-prolines as alkylated dipeptide precursors
A different approach that involves N -alkylated residues to prevent hydrogen bonding is the use of cyclic structures called pseudo-prolines (51). These structures are compounds formed by reaction of serine, threonine, or cysteine residues with formaldehyde, acetaldehyde, or an acetone equivalent. The products are oxazolidines or thiazolidines that bear a carboxyl group and one or more methyl groups on the other carbon atoms of the rings (Fig. 7). The pseudoprolines are employed in synthesis as the Fmoc-dipeptides, with the cyclic structure at the carboxy-terminus. The Fmoc-amino acid is combined with the terminal residue either before or after formation of the ring. The pseudo-proline-containing dipeptide is coupled to the amino group of the peptide that is being assembled. At the end of the synthesis, the pseudo-proline is converted back to its precursor amino acid by acidolysis (Fig. 7). The strength of acid necessary is dictated by the nature of substituents R1, R2 that originate from the carbonyl compound and the hetero atom O or S of the ring. Methyl groups render the cyclic structures more sensitive to acid; the thiazolidine ring is much more stable to acid than the oxazolidine ring.
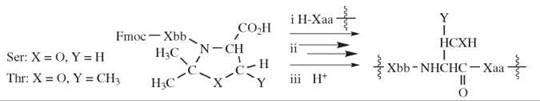
Figure 7. The use of pseudo-prolines. (i) Fmoc-dipeptide containing the pseudo-proline at the COOH-terminus is coupled to a chain, (ii) the chain is extended, and (iii) the original amino-acid residue is regenerated.
Microwave-assisted couplings
The new technology of facilitating chemical reactions by use of microwave energy has been applied to the synthesis of peptides (52, 53). The theory is that the energy excites the growing peptide chain, which prevents aggregation during chain extension. Coupling times are substantially reduced, and products of enhanced purity have been obtained.
Other Peptide-Bond Forming Approaches
A new approach to the synthesis of activated esters of peptides is through alkyl thio esters R(O=C)SR' obtained by solid-phase synthesis (54). The segment Fmoc-Xcc-Xxxx-Xaa-S-CH2 spacer-1-linker-resin is assembled using Boc/Bzl chemistry except for the last residue, thio ester Fmoc-Xcc-Xxxn-Xaa-S-CH2 spacer-H is released by acidolysis with HF at the bond indicated by the arrow and converted into activated peptide Fmoc-Xcc-Xxxn-Xaa-ODhbt by reaction with HODhbt in the presence of silver ion. These esters react with amino components, but side-chain amino groups have to be protected. They are used for chemoselective ligation, in which two unprotected peptides, each possessing a uniquely reactive functional group at one of the two termini, are allowed to combine in denaturing aqueous solvent (55, 56). One type involves reaction of the thio ester with a side-chain sulfur atom at the terminus of the other segment followed by an S-to-N acyl shift.
References
1. V. Glaser, Bioprocessing: market growing for custom-made peptides. Genetic Eng. Biotech. News 2006; 26(13).
2. C. Johansson, L. Blomberg, W. Hlebowicz, H. Nicklasson, B. Nilsson, L. Andersson, Industrial production of an oxytocin antagonist: Synthetic approaches to the development of a multikilogram scale solution synthesis. In: H. L. S. Maia (ed.), Peptides 1994. Proc 23rd Eur. Pept. Symp. Escom Publishers, Leiden, 1995, pp. 34-35.
3. N. L. Benoiton and F. M. F. Chen, 2-Alkoxy-5(4H)-oxazolones from N -alkoxycarbonylamino acids and their implication in carbodiimide-mediated reactions in peptide synthesis. Can. J. Chem. 1981; 59:384-389.
4. N. L. Benoiton and F. M. F. Chen, Sometimes it is neither a racemisation nor an epimerisation but an enantiomerisation. A plea for preciseness in the use of terms describing stereomutations that occur in peptide synthesis. Int. J. Pept. Prot. Res. 1994; 44:399-400.
5. N. L. Benoiton and F. M. F. Chen, Not the alkoxycarbonylamino-acid O-acylisourea. J. Chem. Soc. Chem. Commun. 1981; 543-545.
6. F. Weygand, D. Hoffmann, and E. Wiinsch. Z Naturforsch B 1966; 21:426.
7. W. Konig and R. Geiger, A new method for the synthesis of peptides: Activation of the carboxyl group with dicyclohexylcarbodiimide and 1-hydroxybenzotriazoles. Chem. Ber. 1970; 103:788-798.
8. W. Konig and R. Geiger, A new method for the synthesis of peptides: Activation of the carboxyl group with dicyclohexylcarbodiimide and 3-hydroxy-4-oxo-3,4-dihydro-1.2.3-benzotriazine. Chem. Ber. 1970; 103:2034-2040.
9. L. A. Carpino, 1-Hydroxy-7-azabenzotriazole. An efficient peptide coupling additive. J. Am. Chem. Soc. 1993; 115:4397-4398.
10. Anderson JW, Zimmerman JE, Callahan FM. The use of esters of N-hydroxysuccinimide in peptide synthesis. J. Am. Chem. Soc. 1964; 86:1839-1842.
11. J. C. Sheehan and G. P. Hess, A new method of forming peptide bonds. J. Am. Chem. Soc. 1955; 77:1067-1068.
12. J. Podlech, Carbodiimides. In: M. Goodman, F. Arthur, L. Moroder, and C. Toniolo (eds.), Synthesis of Peptides and Pep- tidomimetics, Houben-Weyl Methods of Organic Chemistry, E 22a. Stuttgart: George Thieme Verlag, 2002, pp. 517-533.
13. T. Miyazawa, Otomatsu, Y. Fukui, T. Yamada, and S. Kuwata, Simultaneous use of 1-hydroxybenzotriazole and copper(II) chloride as additives for racemization-free and efficient synthesis by the carbodiimide method. Int. J. Pept. Prot. Res. 1992; 39:308-314.
14. B. Belleau and G. Malek, A new convenient reagent for peptide syntheses. J. Am. Chem. Soc. 1968; 90:1651.
15. H. Hagenmeier and H. Frank, Increased coupling yields in solid phase peptide synthesis with a modified carbodiimide coupling procedure. Hoppe-Seyler’s Z. Physiol. Chem. 1972; 353: 1973-1976.
16. F. M. F. Chen, K. Kuroda, and N. L. Benoiton, A simple preparation of symmetrical anhydrides of N-alkoxycarbonylamino acids. Synthesis 1978; 928.
17. E. J. Heimer, C. Chang, T. Lambros, and J. Meienhofer, Stable isolated symmetrical anhydrides of N a-9-fluorenylmethyloxy carbonylamino acids in solid-phase peptide synthesis. Int. J. Pept. Prot. Res. 1981; 18:237-241.
18. G. W. Anderson, J. W. Zimmerman, and F. M. Callahan, A reinvestigation of the mixed carbonic anhydride method of peptide synthesis. J. Am. Chem. Soc. 1967; 89:5012-5017.
19. F. M. F. Chen and N. L. Benoiton, The preparation and reactions of mixed anhydrides of N -alkoxycarbonylamino acids. Can. J. Chem. 1987; 65:619-625.
20. N. L. Benoiton, Y. Lee, and F. M. F. Chen, Isopropyl chloroformate as a superior reagent for mixed anhydride generation and couplings in peptide synthesis. Int. J. Pept. Prot. Res. 1988; 31:577-580.
21. B. Castro, J. R. Dormoy, G. Evin, and C. Selve, Peptide coupling reagents IV (1) - Benzotriazole N-oxytrisdimethylamino phosphonium hexafluorophosphate (B.O.P.). Tetrahedron Lett. 1975; 1219-1222.
22. V. Dourtoglou, Ziegler J.-C., and B. Gross, L’hexafluoro phosphate de O-benzotriazolyl-N,N-tetramethyluronium hexafluorophosphate: a new and efficient peptide coupling reagent. (in French) Tetrahedron Lett. 1978; 1269-1272.
23. J. Coste, D. Le-Nguyen, and B. Castro, PyBOP: A new peptide coupling reagent devoid of toxic by-product. Tetrahedron Lett. 1990; 31:205-208.
24. J. Coste, M.-N. Dufour, D. Le-Nguyen, and B. Castro, BOP and congeners: Present status and new developments. In: J. Rivier, R. Garland, and R. Marshall (eds.), Peptides Chemistry, Structure, Biology. Leiden Escom publishers, 1990, pp. 885-888.
25. J. Coste, E. Frerot, P. Jouin, and B. Castro, Oxybenzotriazole free peptide coupling reagents for N-methylated amino acids. Tetrahedron Lett. 1991; 32:1967-1970.
26. I. Abdelmody, F. Albericio, L. A. Carpino, B. M. Foxman, and S. A. Kates, Structural studies of reagents for peptide bond formation: crystal and molecular structures of HBTU and HATU. Lett. Pept. Sc. 1994; 1:57-67.
27. A. Carpino, H. Imazumi, A. El-Faham, F. J. Ferrer, C. Zhang, Y. Lee, B. M. Foxman, P. Henklein, C. Hanay, C. Mugge, H. Wenschuh, The uronium/guanidinium peptide coupling reagents: finally the true uronium salts. Angew. Chem. Int. Edn. 2002; 41:442-445.
28. S. Chen and J. Xu, A new coupling reagent for peptide synthesis. Benzotriazol-yl-bis(pyrrolidino)-carbonium hexafluorophosphate (BCC). Tetrahedron Lett. 1992; 33:647-650.
29. O. Marder and F. Albericio, Industrial application of coupling reagents in peptides. Chimica Oggi 2003; 21:6-11.
30. L. A. Carpino, A. A. Abdel-Maksoud, E. M. E. Mansour, M. A. Zewail. Segment coupling to a highly hindered N-terminal, alamethicin-related a-aminoisobutyric acid (Aib) residue. Tetrahedron Lett. 2007; 48:7404-7407.
31. L. A. Carpino, B. J. Cohen, K. E. Stephens, S. Y. Sadat-Aalaee, J.-H. Tien, and D. C. Lakgridge, (9-Fluorenylmethyl)oxycarbonyl (Fmoc) amino acid chlorides. Synthesis, characterization, and application to the rapid synthesis of short peptide segments. J. Org. Chem. 1986; 51:3732-3734.
32. L. A. Carpino, M. Beyermann, H. Wenschuh, and M. Bienert, Peptide Synthesis via amino acid halides. Acc. Chem. Res. 1997; 29:268-274.
33. E. Falb, T. Yechezkel, Y. Salitra, and C. Gilon, In situ generation of Fmoc-amino acid chlorides using bis-(trichloromethyl) carbonate and its utilization for difficult couplings in solid-phase peptide synthesis. J. Pept. Res. 1998; 53:507-517.
34. H. N. Gopi and V. V. Suresh Babu Zinc-promoted simple synthesis of oligomer-free Na-Fmoc-amino acids using Fmoc-Cl as an acylating agent under neutral conditions. J. Pept. Res. 2000; 55:295-299.
35. J. Lutz, H.-J. Musiol, L. Moroder, Acyl azides. In: M. Goodman, F. Arthur, L. Moroder, and C. Toniolo (eds.), Synthesis of Peptides and Peptidomimetics, Houben-Weyl Methods of Organic Chemistry, E 22a, Stuttgart: George Thieme Verlag, 2002, pp. 427-441.
36. T. Shiori, T. Ninomia, and S. Yamada, Diphenylphosphoryl azide. A new convenient reagent for a modified Curtius reaction and for peptide synthesis. J. Am. Chem. Soc. 1972; 94:6203-6205.
37. J. Honzl and J. Rudinger, Amino acids and peptides. XXXIII. Nitrosyl chloride and butyl nitrite as reagents in peptide synthesis by the azide method; suppression of amide formation. Coll. Czech. Chem. Commun. 1961; 26:2333-2344.
38. J. K. Chang, M. Shimuzu, and S.-S. Wang, Fully automated synthesis of fully protected peptide hydrazides on recycling hydroxymethyl resin. J. Org. Chem. 1976; 41:3255-3258.
39. P. Wang, R. Layfield, R. J. Mayer, and R. Ramage, Transfer active ester condensation: a novel technique for peptide segment coupling. Tetrahedron Lett. 1998; 39:8711-8714.
40. N. L. Benoiton, Active esters. In: Synthesis of Peptides and Peptidomimetics, Houben-Weyl Methods of Organic Chemistry, E 22a, M. Goodman, F. Arthur, L. Moroder, and C. Toniolo (eds.), Stuttgart: George Thieme Verlag, 2002, pp. 443-474.
41. J. H. Jones and G. T. Young, Anchimeric acceleration of aminolysis of esters and its application to peptide synthesis. Chem. Commun. 1967: 35-36.
42. H. Gross and L. Bilk, On the reaction of N-hydroxysuccinimide with dicyclohexylcarbodiimide. Tetrahedron 1968; 24:6935-6594.
43. Z. J. Kaminski, B. Kolesinska, J. Kolesinska, G. Sabatino, M. Chelli, P. Rovero, M. Blaszczyk, M. L. Glowka, A. M. Papina. N-Triazinylammonium tetrafluoroborates. A new generation of coupling reagents usefull for peptide synthesis. J. Am. Chem. Soc. 2005; 127:16912-16920.
44. W. D. Fuller, M. S. Verlander, and M. Goodman, A procedure for the facile synthesis of amino-acid N-carboxyanhydrides. Biopolymers 1976; 15:1869-1871.
45. R. Hirschmann, H. Schwam, R. G. Strachan, E. F. Schoenewaldt, H. Barkemeyer, S. M. Miller, J. B. Conn, V. Garsky, D. F. Veber, and R. G. Denkewalter, The controlled synthesis of peptides in aqueous medium. The preparation and use of novel a-amino acid N-carboxyanhydrides. J. Am. Chem. Soc. 1971; 93:2746-2754.
46. W. D. Fuller, M. P. Cohen, M. Shabankareh, R. K. Blair, M. Goodman, and F. R. Naider, Urethane-protected amino acid N-carboxyanhydrides and their use in peptide synthesis. J. Am. Chem. Soc. 1990; 112:7414-7416.
47. W. S. Hancock, D. J. Prescott, P. R. Vagelos, and G. R. Marshall, Solvation of the polymer matrix. Source of truncated and deletion sequences in solid phase synthesis. J. Org. Chem. 1973; 38:774-781.
48. C. Hyde, T. Johnson, D. Owen, M. Quibell, R. C. Sheppard Some ‘difficult sequences’ made easy. A study of interchain association in solid-phase peptide synthesis. Int. J. Pept. Prot. Res. 1994; 43:431-440.
49. Y. Sohma, M. Sasaki, Y. Hayashi, T. Kimura, and Y. Kiso, Novel and efficient synthesis of difficult sequence-containing peptides through O -N intramolecular acyl migration reaction of O-acyl isopeptides. Chem. Commun. 2004; 124-125.
50. T. Johnson, M. Quibell, and R. C. Sheppard, N, O-bisFmoc derivatives of N-(2-hydroxy-4-methoxybenzyl)-amino acids: useful intermediates in peptide synthesis. J. Pept. Sci. 1995; 1:11.
51. T. Wohr, F. Wahl, A. Nefzi, B. Rodwedder, T. Sato, X. Sun, and M. Mutter, Pseudo-prolines as a solubilizing, structure-disrupting protection technique in peptide synthesis. J. Am. Chem. Soc. 1996; 118:9218.
52. H.-M. Yu, S.-T. Chen, and K.-T. Wang, Enhanced coupling efficiency in solid-phase peptide synthesis by microwave irradiation. J. Org. Chem. 1992; 57:4781-4784.
53. M. Erdelyi and A. Gogoll, Rapid microwave-assisted solid phase peptide synthesis. Synthesis 2002; 11:1592-1596.
54. S. Aimoto, Polypeptide synthesis by the thioester method. Biopolymers 1999; 51:247-265.
55. T. W. Muir, P. E. Dawson, and S. B. H. Kent, Protein synthesis by chemical ligation of unprotected peptides in aqueous solution. Methods Enzymol. 1997; 289:266-298.
56. J. P. Tam, A. Yu, and A. Miao, Orthogonal ligation strategies for peptide and protein synthesis. Biopolymers 1999; 51:311-332.
Further Reading
F. Albericio and L. A. Carpino, Coupling reagents and activation. Methods Enzymol. 1997; 289:104-126.
N. L. Benoiton, Chemistry of Peptide Synthesis. Boca Raton, FL: Taylor & Francis, 2005.
E. Gross and J. Meienhofer, eds., The Peptides: Analysis, Synthesis, Biology, vol 1. New York: Academic Press, 1979.
See Also
Amino Acids, Chemical Properties of
Click Peptides, Design and Application of
Peptide Combinatorial Libraries
Solid-Phase Synthesis of Biomolecules