CHEMICAL BIOLOGY
Cyclooxygenase Inhibition, Recent Advances in the Mechanisms of
Melissa V. Turman, Anna L. Blobaum, Mary E. Konkle and Lawrence J. Marnett, A.B. Hancock Jr. Memorial Laboratory for Cancer Research, Departments of Biochemistry, Chemistry, and Pharmacology, Vanderbilt Institute of Chemical Biology, Center in Molecular Toxicology, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, Tennessee
doi: 10.1002/9780470048672.wecb108
Cyclooxygenases (COXs) are key enzymes of bioactive lipid metabolism that catalyze the committed step in prostaglandin biosynthesis. Inhibition of COX catalysis is a principal mode of action of nonsteroidal antiinflammatory drugs (NSAIDs). The combination of structural and functional analysis of COX-inhibitor interactions has provided important insights into the molecular determinants of inhibition. Although COX protein structure does not change significantly on inhibitor binding, a surprising variety of binding modes and molecular interactions are observed with different NSAIDs. This article focuses on recent advances in our understanding of these interactions with emphasis on arylcarboxylic acid-based inhibitors and their derivatives.
Cyclooxygenases (COX-1 and COX-2) are bifunctional enzymes that carry out two sequential reactions in spatially distinct but mechanistically coupled active sites—the double dioxygenation of arachidonic acid to prostaglandin G2 (PGG2) and the reduction of PGG2 to PGH2 (Eq. 1) (1). Arachidonic acid oxygenation occurs in the cyclooxygenase (COX) active site, and PGG2 reduction occurs in the peroxidase (POX) active site. The COXs are homodimers of 70 kDa subunits, and dimerization is required for structural integrity and catalytic activity (2). The first crystal structure solved for a COX enzyme (ovine COX-1 with the bound inhibitor flurbiprofen) revealed an asymmetric unit containing two identical monomers that exhibit extensive contacts in a large subunit interface; each subunit contains the inhibitor bound only in the COX active site (3). Although it has been assumed that both subunits are active simultaneously, recent work suggests that substrate or inhibitor binding in the COX active site at one subunit precludes the binding of another molecule at the other subunit (4). This cooperativity between subunits is consistent with the observation that COX-2 dimers only bind a single molecule of flurbiprofen tightly, and this is sufficient to inhibit all COX activity (4). The molecular details of the communication between subunits are not yet understood, but their elucidation will have a significant impact on defining COX-inhibitor interactions.

Ligand Binding to COX Enzymes
Each monomer of COX consists of three structural domains: a short N-terminal epidermal growth factor domain, a membranebinding domain, and a large, globular C-terminal catalytic domain (Fig. 1a) (3). The COX and POX active sites are located on opposite sides of the catalytic domain with the heme prosthetic group positioned at the base of the peroxidase site. The epidermal growth factor domain and catalytic domain create the dimer interface and place the two membrane-binding domains on the same face of the homodimer about 25 A apart (5, 6). The membrane-binding domain of cyclooxygenase is composed of four amphipathic alpha helices, with hydrophobic and aromatic residues that project from the helices to create a surface that interacts with one face of the lipid bilayer (3). Three of the four helices lie in the same plane, whereas the last helix (helix D) projects up into the catalytic domain (5). The catalytic domain constitutes the majority of the COX monomer and is the site of substrate binding and NSAID action.
Substrate and inhibitor gain access to the COX active site (Fig. 1b) at the base of the membrane-binding domain, which leads into a long hydrophobic channel that extends deep into the catalytic domain interior (3). This hydrophobic channel narrows at the interface between the membrane-binding domain and the catalytic domain to form a constriction composed of three residues (Arg-120, Tyr-355, and Glu-524) that separate the “lobby” from the active site. The active sites of COX-1 and COX-2 are nearly identical in the amino acid residues that constitute the primary shell of the active site but differ in the presence of a side pocket in COX-2 bordered by Val-523 (isoleucine in COX-1) that is located above the constriction site (7, 8). The solvent-accessible surface available in the COX-2 active site is larger than that in COX-1 because of one Val-523 to Ile substitution in the primary shell and several key substitutions in the secondary shell (Arg-513 to His and Val-434 to Ile) (7). The single Val-523 to Ile substitution in the primary shell and the key substitutions in the secondary shell confer selectivity for the diarylheterocycle class of COX-2 selective inhibitors (e.g., celecoxib and rofecoxib). In addition, the last helix of the membrane-binding domain (helix D) is positioned differently in COX-2 and shifts the location of Arg-120 at the constriction site, which allows for a larger solvent-accessible surface at the interface between the membrane-binding domain and the COX active site in COX-2.
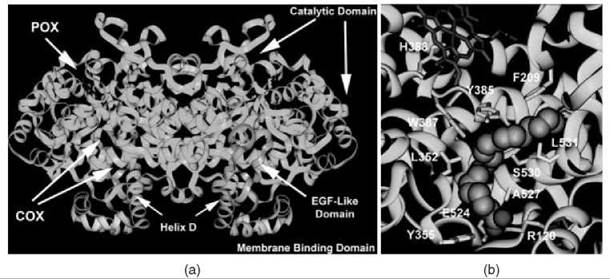
Figure 1. (a) Structural representation of the murine COX-2 dimer. The N-terminal epidermal growth factor domain is designated in pink and leads into the four a-helices of the membrane binding domain (yellow). Helix D projects up into the COX active site, which is located at the base of the large, globular catalytic domain (cyan). The heme prosthetic group (red) lies in the POX active site. (b) Arachidonic acid bound in the active site of COX-1. The carboxylate of the substrate ion-pairs with Arg-120 and hydrogen-bonds with Tyr-355 at the constriction site, projects up the hydrophobic channel, and makes an L-shaped bend around Tyr-385. The heme prosthetic group is designated in red. Residues that contact arachidonic acid in the active site channel are shown in yellow.
Time-Dependent Inhibition of COX by Indomethacin and its Ethanolamides
Kinetic studies of COX inhibition reveal that many NSAIDs interact with COX through a multistep mechanism in which a rapid, reversible step is followed by one or more slow steps that may be poorly reversible (Eq. 2) (9, 10):
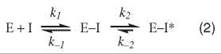
Although dissociation of the E-I* complex is often too small to be measured, none of the NSAIDs other than aspirin cause covalent modification of COX enzymes. Rather, the final step of binding for many NSAIDs corresponds to the formation of a tightly bound and functionally irreversible protein-inhibitor complex, E-I*. Indomethacin (INDO) is a highly potent, time-dependent inhibitor of COX (10). Although its time-dependent kinetics were first reported over 30 years ago, the mechanism of its tight binding was not understood. A crystal structure of the complex of INDO with murine COX-2 provides a detailed view of the binding orientation and critical protein-ligand interactions (8). The carboxylic acid of INDO binds at the constriction site of COX, forming an ionic bond with Arg-120. The heterocyclic scaffold occupies the oxygenase channel, with the methoxy group projecting toward Val-523 and the para-chlorobenzoyl moiety projecting toward Tyr-385 and Trp-387. A small, hydrophobic pocket formed by Val-349, Ala-527, Ser-530, and Leu-531 surrounds the 2'-methyl group on the indole ring (Fig. 2a).
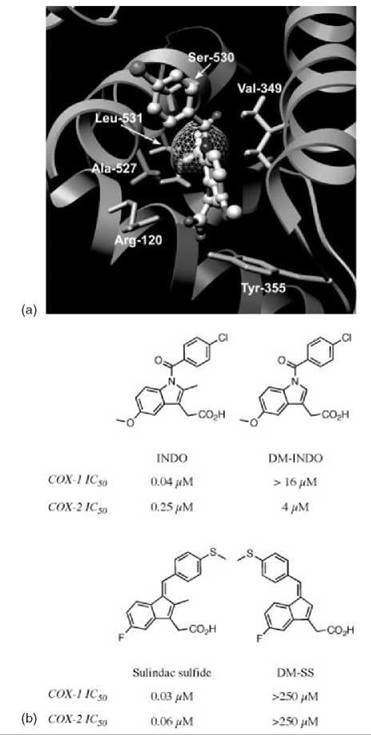
Figure 2. Importance of the 2'-methyl group for inhibition of COX by indomethacin and sulindac sulfide. (a) Indomethacin bound in the active site of COX-2. The 2'-methyl group of indomethacin, shown with its Van der Waals surface, sits in a hydrophobic pocket that consists of Val-349, Ala-527, Ser-530, and Leu-531. Hydrogens are included on the residues of the hydrophobic pocket and the 2'-methyl group of indomethacin. (b) Structures and inhibitory activities of indomethacin, sulindac sulfide, and their des-methyl analogs.
The importance of the interaction with the 2'-methyl group was revealed by a combination of protein mutagenesis and inhibitor modification (11). Site-directed mutants of COX-2 were generated, which altered the size of the hydrophobic pocket via substitution of Val-349 by Ala, Ile, and Leu. Enlarging the size of the pocket (V349A) did not significantly alter the potency or binding kinetics of INDO. Although INDO exhibited diminished potency toward V349I, the binding kinetics for V349I were similar to the wild-type COX-2 and V349A. However, mutation of Val-349 to Leu seems to block access to the hydrophobic pocket in an efficient manner. The potency of INDO inhibition of V349L was reduced 50-fold when compared with wild-type COX-2. Even more striking is the difference in kinetics displayed by V349L. The values of k2, the forward rate constant for formation of the tightly bound complex E—I*, were similar for wild-type COX-2 and all Val-349 mutants. Dissociation of this complex, as judged by k—2, is not measurable for wild-type COX-2, V349A, or V349I. However, a measurable and, in fact, rapid k—2 was observed with V349L. As a result, most of the inhibitor was competed off within a few minutes after the addition of arachidonic acid. This finding suggests that the reversibility of the second step in the time-dependent mechanism is the primary determinant of the potency of COX inhibition by INDO and that this is largely defined by interactions between the 2'-methyl group and the hydrophobic pocket.
A derivative of INDO that lacks the 2'-methyl group was synthesized, and its interactions with COX-1 and COX-2 were studied (Fig. 2b) (11). This compound, des-methyl INDO (DM-INDO), is a poor inhibitor of wild-type COX-1 and COX-2 and all Val-349 COX-2 mutants. Again, potency is dependent on the reversibility of the second step of the binding mechanism. Curiously, DM-INDO displays COX-2 selectivity, despite complete sequence conservation within the hydrophobic pocket in COX-1 and COX-2. Although selectivity may be related to the magnitude of k—2, which is an order of magnitude faster for COX-1 than for COX-2, the structural basis of selectivity of DM-INDO inhibition has not been elucidated.
Sulindac sulfide, the bioactive metabolite of sulindac, is structurally very similar to INDO and is a slow, tight-binding inhibitor of COX (Fig. 2b) (12, 13). As with INDO, removal of the methyl group from sulindac sulfide results in loss of COX-1 and COX-2 inhibition (14). However, it should be noted that the benzylidine double bond of des -methyl sulindac sulfide (DM-SS) exists in the E-conformation, whereas sulindac sulfide exists in the Z-conformer.
Many clinically relevant NSAIDs exert off-target effects unrelated to their ability to inhibit COX enzymes. For example, INDO and SS induce apoptosis of tumor cells and modulate y-secretase activity (15, 16). INDO also activates the nuclear transcription factor PPARy (17). The complexity of in vivo pharmacologic effects makes it a challenge to separate the contribution of COX inhibition from other effects in a given pharmacologic response. Thus, the removal of COX inhibitory activity by a minor modification, such as the removal of a methyl group, provides an opportunity to dissect COX-dependent and COX-independent effects of certain NSAIDs. In fact, DM-INDO and DM-SS activate PPARy in HCA-7 cells with dose responses similar to those of the parent drugs (14). Likewise, the des -methyl compounds exhibit potency similar to the parent compounds in their ability to induce apoptosis in RKO cells, a human colon cancer cell line, and to activate PPARy in cellular reporter assays.
Gastrointestinal toxicity is a classic side effect of INDO and other potent, nonselective COX inhibitors. It is widely accepted that this toxicity arises from inhibition of PGE2 production in the gastric mucosa; however, other mechanisms have been proposed (15, 18). C57BL6 mice were administered INDO or DM-INDO at 5 mg/kg, a dose above the LD50 of INDO. Whereas the gastric mucosa of INDO-treated animals exhibited significant tissue necrosis, the gastric tissue from DM-INDO-treated animals was identical to that of healthy control animals (14). Furthermore, the LD50 of DM-INDO in C57BL6 mice was 20-fold higher than that of INDO, which suggests that toxicity of INDO is closely associated with inhibition of COX.
Understanding how a drug works on the molecular level is critical to elucidating its pharmacologic effects. INDO provides a striking case study of how subtle interactions, revealed by protein structure-function analysis, can be exploited to dissect the off-target effects of drugs. DM-INDO and DM-SS may provide scaffolds for probing or fine-tuning the beneficial side activities of the parent drugs, while reducing dose-limiting toxicity.
For many NSAIDs, including flurbiprofen and INDO, the ionic bond between Arg-120 and the carboxylic acid of the inhibitor is absolutely required for time-dependent inhibition of COX-1 (10, 19); the methyl esters of INDO and other NSAIDs were shown to be very poor inhibitors of COX-1 (10). However, this interaction is not a universal requirement for inhibition of COX-2 (20). Conversion of the INDO carboxy- late group to neutral esters and amides provides a general and facile method for generating COX-2-selective inhibitors (21). A notable exception is a series of a-substituted ethanolamides of INDO, which exist as R/S -enantiomeric pairs (22). Across a range of a-substitutions, the R-enantiomers are consistently COX-2-selective. However, the S -enantiomers efficiently inhibit both COX-1 and COX-2.
Crystal structures of the pair of a-ethyl-substituted enantiomers bound to COX-1 provide a surprising structural basis for the enantioselectivity of inhibition (23). The R-enantiomer (8) binds in a conformation similar to that of INDO bound to COX-1 or COX-2 (Fig. 3). The para-chlorobenzoyl group is oriented toward Tyr-385 and Trp-387, the methoxy projects toward the side pocket, and the ethanolamide is positioned at the constriction site. The hydroxyl group of the ethanolamide forms a hydrogen bond to the guanidinium of Arg-120. To accommodate the hydroxyethyl substituent, Arg-120 must rotate away from Tyr-355 and toward Glu-524. The ethyl group extends through the constriction.
The S -enantiomer (9) adopts a very different conformation (Fig. 3). In this case, the methoxy group projects toward the apex of the channel, with the para-chlorobenzoyl wedged into a groove below Leu-531 and the 2'-methyl group positioned above Tyr-355. The ethanolamide moiety inserts into the side pocket, which consists of His-90, Gln-192, Leu-517, Phe-518, and Ile-523. The substituted ethanolamide of 9 makes several hydrophobic interactions with Phe-518 and Ile-523. The hydroxyl group can hydrogen bond to His-90 and Gln-192. In COX-2, the sulfone or sulfonamide moiety of the diarylheterocyles use this side pocket, which is enlarged by conservative amino acid substitutions in the primary and secondary shells, and provide the basis for COX-2-selectivity of this class of inhibitors (7, 8). Notably, attempts to model 8 in a conformation similar to the noncanonical conformation of 9 indicate that the R-enantiomer cannot take advantage of the same network of interactions and could not avoid steric clashes with protein side chains.
The kinetics of 8 and 9 binding and inhibition with COX-1 reveal an interesting profile. Pre-steady state kinetic measurements indicate that binding of 8 and 9 proceeds by a multistep mechanism like that of INDO (Eq. 2). Although KI, the ratio of k—1 to k1, is similar for 8, 9, and INDO, formation of E—I* proceeds at a much faster rate (10-fold) for 8 and 9 than for INDO (11, 23). To attain maximal inhibition in vitro, INDO must be preincubated with COX for at least 10 minutes before the addition of substrate (9). However, maximal inhibition of COX-1 by 8 and 9 can be achieved by a 30 second preincubation with the protein (23). The potency of inhibition once again is determined by the dissociation of the E—I* complex. As with INDO, k—2 could not be detected for 9, and substrate cannot compete efficiently with INDO or 9 for the oxygenase site, even after prolonged incubations. In contrast, k—2 for 8 is measurable and of the same order of magnitude as k2. Even when COX-1 is preincubated with 8, most enzymatic activity is recovered following incubation with substrate.

Figure 3. Structural basis of COX inhibition by α-substituted indomethacin ethanolamides. Structures and inhibitory activities of indomethacin and the a-ethyl indomethacin ethanolamides are shown. The crystal structures of indomethacin bound to COX-2 and 8 and 9 bound to COX-1 reveal differences in the binding of the R- and S-enantiomers to COX-1.
These results demonstrate that the side pocket of COX-1, which has been considered sterically inaccessible, can bind certain (S)-hydroxyethylamide derivatives of INDO. This study also offers insight into the selective binding of amide derivatives of INDO to COX-2. It seems that the amides associate with COX-1 and COX-2 but only bind tightly to COX-2. If all INDO-amides adopt a conformation in both enzymes analogous to that of the (R)-hydroxyethylamide 8, then this complex is only stable in COX-2. Because the orientation of Arg-120 is altered in the COX-1-compound 8 complex relative to other COX-1 inhibitor complexes, it may be that COX-2 can better accommodate this conformation than COX-1. It will be interesting to test this hypothesis and to identify the protein determinants responsible for the differential stability.
Inhibition by Aspirin and Salicylate
The development of aspirin in the late 1890s is a landmark in the evolution of the pharmaceutical industry (24). Many decades later, aspirin was shown to inhibit prostaglandin biosynthesis by covalently modifying COX (25, 26). Inactivation results from acetylation of Ser-530, which is located above Arg-120 and across from Tyr-385 in the COX active site (27). Mutagenesis of Tyr-385 to Phe in COX-2 reduces acetylation of Ser-530 by greater than 90%, which suggests the importance of hydrogen-bond formation in acetylation (28). Indeed, a crystal structure of COX-1 acetylated by bromoacetylsalicylic acid reveals a hydrogen bond between Tyr-385 and the carbonyl oxygen of the bromoacetyl group (29). It has been proposed that hydrogen bonding by Tyr-385 stabilizes the incipient anion of the transition state during acetylation of Ser-530 (Fig. 4) (28).

Figure 4. Putative mechanism of aspirin acetylation of COX enzymes. Arg-120 positions aspirin adjacent to Ser-530 by ion-pairing; then Tyr-385 stabilizes the transition state for acetylation by hydrogen-bonding to the developing oxyanion.
Tyr-385 is a critical residue for cyclooxygenase catalysis. Reaction of the heme prosthetic group with a hydroperoxide (e.g., peroxynitrous acid or PGG2) generates a higher oxidation state of the heme that oxidizes Tyr-385 to a tyrosyl radical (30). This radical oxidizes arachidonic acid in the first step of cyclooxygenase catalysis. At the end of the catalytic cycle, the tyrosyl radical is regenerated. Thus, Tyr-385 only exists in the fully covalent state in the resting, catalytically inactive enzyme but is oxidized to a tyrosyl radical in the active enzyme. This finding may have implications for the ability of NSAIDs to interact with the enzyme if the conformation of the active site residues changes during catalysis. For example, treatment of COX enzymes with fatty acid hydroperoxides reduces the potency of salicylate and acetaminophen as inhibitors (31, 32). This reduction seems to be caused by the peroxide oxidation of the heme prosthetic group (and presumably the oxidation of Tyr-385) because substitution of Mn3+-protoporphyrin IX for heme eliminates hydroperoxide antagonism of salicylate and acetaminophen inhibition (31, 32). Hydroperoxide-antagonism of COX inhibition may be related to the purported differential sensitivity of COX-1 and COX-2 to certain NSAIDs because COX-1 requires approximately a 10-fold higher concentration of peroxide than COX-2 to achieve full activation (33, 34).
Acetylation of COX-1 completely abolishes cyclooxygenase activity presumably by blocking the binding of arachidonic acid in the COX active site. However, acetylated COX-2 can oxidize arachidonic acid to a 15(R)-hydroperoxy derivative (35). This ability is because of the greater size of the COX-2 active site. Mutagenesis of the side pocket residues, Val-523, Arg-513, and Val-434, to the COX-1 variants (Ile-523, His-513, and Ile-434) eliminates the ability of acetylated COX-2 to oxidize arachidonic acid (36). This finding indicates that acetylated COX-2 can accommodate arachidonic acid in its active site in a binding mode that allows oxygenation at carbon-15 to 15-hydroperoxyeicosatetraenoic acid. This finding may be pharmacologically important because 15-hydroperoxyeicosate- traenoic acid is a substrate for oxygenation by lipoxygenases to lipoxin A4 (37). Lipoxin A4 and related compounds have potent antiinflammatory activity, and the contribution to its formation by acetylated COX-2 may account for some of the antiinflammatory activity exhibited by aspirin.
Diclofenac and Lumiracoxib: Novel Mechanisms for Inhibition and COX-2 Selectivity
The importance of Tyr-385 in aspirin acetylation of Ser-530 suggests that it may play a broader role in interacting with electron-rich centers. In fact, the crystal structure of a catalytically inactive mutant of COX-2 with bound arachidonic acid reveals the arachidonate bound in an inverted, noncanonical orientation in which its carboxylate group is hydrogen-bonded to both Tyr-385 and Ser-530 (38). This conformation is not a productive conformation for arachidonate oxygenation, but it provides insight into the binding of COX-2 (and possibly COX-1) to other alkyl or aryl carboxylic acids.
Many crystal structures are available of COX-1 or COX-2 with bound carboxylic acid-containing inhibitors, and for the most part the inhibitors are bound with their carboxylates ion-paired to Arg-120 and hydrogen-bonded to Tyr-355 at the base of the cyclooxygenase active site (3, 8). However, a crystal structure of diclofenac bound in the active site of COX-2 (Fig. 5) demonstrates an inverted binding mode of the inhibitor with its carboxylic acid moiety coordinated to Ser-530 and Tyr-385 at the top of the active site and not ion-paired nor hydrogen-bonded to Arg-120 and Tyr-355 (39). This binding mode seems to be the binding mode responsible for inhibition because mutagenesis of Ser-530 to Ala abolishes diclofenac inhibition whereas mutagenesis of Arg-120 to Gln is without effect (39). Interestingly, diclofenac also seems to make use of the small hydrophobic binding pocket composed of Ala-527, Val-349, Ser-530, and Leu-531 that INDO uses to insert its 2'-methyl group. Mutations of Val-349 in this pocket to alanine or leucine alter the size of the pocket and lead to changes in the potency of indomethacin and diclofenac (11, 40).
A close structural analog of the non-selective COX inhibitor diclofenac, lumiracoxib displays a 500-fold greater selectivity for COX-2 than COX-1 in vivo and exhibits a unique pharmacologic profile that includes rapid absorbance and a relatively short plasma half-life (41, 42). Lumiracoxib lacks the tricyclic structure of the diarylheterocycle class of COX-2 selective inhibitors (e.g., celecoxib and rofecoxib) and does not contain a sulfonamide or sulfone group. Although structurally related, lumiracoxib and diclofenac exhibit large differences in the selectivity of COX-2 inhibition, and the molecular basis for this difference in the selectivity of COX inhibition only recently was elucidated.

Figure 5. Crystal structure of diclofenac bound in the active site of COX-2. The carboxyl group of diclofenac is chelated by Tyr-385 and Ser-530 through hydrogen bonding. The structures of diclofenac and lumiracoxib are shown below. The 5'-methyl group of lumiracoxib is a determinant of COX-2 selectivity, and the dihaloaryl group determines the potency of binding.
As expected from its structural resemblance to diclofenac, lumiracoxib binds to COX-2 in an inverted orientation similar to that of diclofenac with hydrogen-bonding interactions between the carboxylate of the inhibitor and Ser-530/Tyr-385 at the top of the active site (43). A comparison of this crystal structure with a model of lumiracoxib bound to COX-1 leads to the conclusion that the COX-2 selectivity of lumiracoxib arises from the insertion of the methyl group on the phenylacetic acid ring of the inhibitor into a small groove provided by the movement of a primary shell leucine residue (Leu-384) in the COX-2 active site. The movement of Leu-384 is thought to be restricted in the active site of COX-1 because of the presence of bulky secondary shell residues lying behind it (Ile-525 and Phe-503) that prevent the movement of Leu-384 with inhibitor bound.
Prior structure-activity studies with diclofenac analogs indicate that methyl or chlorine substituents on the lower aniline ring of the inhibitor in the ortho position are required for potent inhibition of COX (44). Analogs with halogen substitutions (fluorine or chlorine) at the 5' position of the phenylacetic acid ring demonstrate an even higher potency for COX inhibition (44).
Although the selectivity of lumiracoxib for COX-2 has long been demonstrated in vivo, the chemical and structural basis for the balance that exists between potency and COX-2 selectivity was determined only recently for this inhibitor (40). An extensive structure-activity analysis with a library of lu- miracoxib analogs indicates that the 5'-methyl group on the phenylacetic acid ring of the inhibitor is the major determinant for COX-2 selectivity and that the chemical nature of the substituents in the ortho positions on the lower aniline ring exert the major influence on the potency of COX inhibition (2,6-dichloro, 2,6-dimethyl, or 2-chloro-6-methyl substitutions are preferred). Interestingly, the structure-activity study also suggests a contributory role for the chlorine atom of lumiracoxib in COX-2 selectivity, a key finding not divulged by the crystal structure determination. Site-directed mutagenesis of the small hydrophobic binding pocket valine residue (Val-349 to Ala, Ile, or Leu) in COX-2 also supports a role for lumiracoxib binding interactions in this region of the active site (40). The resultant structure-activity analysis of lumiracoxib analogs with the V349 mutants indicates a preference for a chlorine substituent on the lower aniline ring for potent inhibition of COX-2, with F-Cl, Cl-Cl, or Cl-CH3 substitutions allowed at the 2,6 position to maintain potency. These data suggest that the chlorine atom on lumiracoxib (similar to diclofenac) binds in the small hydrophobic pocket that consists of V349, S530, A527, and L531 and that this interaction may contribute to the potent inhibition of COX.
The structure-activity studies with diclofenac and lumiracoxib reveal that the COX-2 inhibitory activity of lumiracoxib results from a fine balance between potency and selectivity. The principal determinant of potency is the dihaloarylamine ring, whereas the metamethylarylacetic acid ring primarily controls selectivity. An F→Cl substitution in the dihaloarylamine ring of lumiracoxib increases the tightness of binding (potency of inhibition) but also reduces selectivity for COX-2. Introduction of the metaethyl group in the arylacetic acid ring enhances COX-2 selectivity but reduces potency. In kinetic analyses, this reduction in potency is manifest by a measurable reverse rate constant (k-2) that allows detectable inhibitor exchange with substrate, especially at elevated concentrations of arachidonic acid. This behavior differs significantly from potent slow, tight-binding NSAIDs, such as indomethacin, sulindac sulfide, and diclofenac, and the COX-2-selective diarylheterocycles, ro- fecoxib and celecoxib. Despite its reduced potency of COX-2 inhibition compared with other NSAIDs and COXIBs, lumiracoxib is the most selective COX-2 inhibitor in vivo. This finding illustrates that multiple factors (such as inhibitor-protein interactions and kinetics of inhibitor association and dissociation) control the selectivity and potency of inhibition in vivo.
The interactions of a variety of selective and nonselective inhibitors with COX-1 and COX-2 have been probed by a combination of crystallography, mutagenesis, and chemical modification. Although the macromolecular protein structure is essentially the same in all COX-inhibitor complexes, seemingly understated differences exist in side-chain conformations, displacement of main-chain protein atoms, and kinetics of inhibitor binding. Different inhibitors use different sets of interactions to optimize their binding in the COX active site, which can afford significant perturbations of selectivity and potency (45). This situation gives rise to a striking variety of bound orientations that illustrate the impact of subtle interactions on ligand-protein association. Problems remain to be solved with this fascinating class of enzymes that should provide additional insights into the potency and selectivity of different classes of inhibitors. The challenge will be to use this information to create novel classes of inhibitors or to address the pharmacologic deficiencies of extant inhibitors (e.g., gastrointestinal toxicity, cardiovascular toxicity, and renal toxicity).
References
1. Rouzer CA, Marnett LJ. Mechanism of free radical oxygenation of polyunsaturated fatty acids by cyclooxygenases. Chem. Rev. 2003; 103:2239-2304.
2. Xiao G, Chen W, Kulmacz RJ. Comparison of structural stabilities of prostaglandin H synthase-1 and -2. J. Biol. Chem. 1998; 273:6801-6811.
3. Picot D, Loll PJ, Garavito RM. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature 1994; 367:243-249.
4. Yuan C, Rieke CJ, Rimon G, Wingerd BA, Smith WL. Partnering between monomers of cyclooxygenase-2 homodimers. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:6142-6147.
5. Garavito RM, Mulichak AM. The structure of mammalian cyclooxygenases. Annu. Rev. Biophys. Biomol. Struct. 2003; 32:183-206.
6. Garavito RM, Mulichak AM. The structure of mammalian cyclooxygenases. Annu. Rev. Biophys. Biomol. Struct. 2003; 32:183-206.
7. Luong C, Miller A, Barnett J, Chow J, Ramesha C, Browner MF. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nature Struct. Biol. 1996; 3:927-933.
8. Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC, Stallings WC. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996; 384:644-648.
9. Smith WL, Lands WEM. Stimulation and blockade of prostaglandin biosynthesis. J. Biol. Chem. 1971; 246:6700-6704.
10. Rome LH, Lands WEM. Structural requirements for time-dependent inhibition of prostaglandin biosynthesis by anti-inflammatory drugs. Proc. Natl. Acad. Sci. U.S.A. 1975; 72:4863-4865.
11. Prusakiewicz JJ, Felts AS, Mackenzie BS, Marnett LJ. Molecular basis of the time-dependent inhibition of cyclooxygenases by indomethacin. Biochemistry 2004; 43:15439-15445.
12. Duggan DE, Hooke KF, Risley EA, Shen TY, Van Arman CG Identification of the biologically active form of sulindac. J. Pharmacol. Exp. Ther. 1977; 201:8-13.
13. Meade EA, Smith WL, DeWitt DL. Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem. 1993; 268:6610-6614.
14. Felts AS, Ji C, Stafford JB, Crews BC, Kingsley PJ, Rouzer CA, Washington MK, Subbaramaiah K, Siegel BS, Young SM, Dannenberg AJ, Marnett LJ. Desmethyl derivatives of indomethacin and sulindac as probes for cyclooxygenase-dependent biology. ACS Chem. Biol. 2007; 2:479-483.
15. Tsutsumi S, Gotoh T, Tomisato W, Mima S, Hoshino T, Hwang HJ, Takenaka H, Tsuchiya T, Mori M, Mizushima T. Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ. 2004; 11:1009-1016.
16. Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 2001; 414:212-216.
17. Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem. 1997; 272:3406-3410.
18. Kim KS, Yoon JH, Kim JK, Baek SJ, Eling TE, Lee WJ, Ryu JH, Lee JG, Lee JH, Yoo JB. Cyclooxygenase inhibitors induce apoptosis in oral cavity cancer cells by increased expression of nonsteroidal anti-inflammatory drug-activated gene. Biochem. Biophys. Res. Commun. 2004; 325:1298-1303.
19. Bhattacharyya DK, Lecomte M, Rieke CJ, Garavito RM, Smith WL. Involvement of arginine 120, glutamate 524, and tyrosine 355 in the binding of arachidonate and 2-phenylpropionic acid inhibitors to the cyclooxygenase active site of ovine prostaglandin endoperoxide H synthase-1. J. Biol. Chem. 1996; 271:2179-2184.
20. Greig GM, Francis DA, Falgueyret JP, Ouellet M, Percival MD, Roy P, Bayly C, Mancini JA, O’Neill GP. The interaction of arginine 106 of human prostaglandin G/H synthase-2 with inhibitors is not a universal component of inhibition mediated by nonsteroidal anti-inflammatory drugs. Mol. Pharmacol. 1997; 52:829-838.
21. Kalgutkar AS, Crews BC, Rowlinson SW, Marnett AB, Kozak KR, Remmel RP, Marnett LJ. Biochemically based design of cyclooxygenase-2 (COX-2) inhibitors: facile conversion of nonsteroidal antiiflammatory drugs to potent and highly selective COX-2 inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:925-930.
22. K.R. Kozak, J.J. Prusakiewicz, S.W. Rowlinson, L.J. Marnett Enantiospecific, selective cyclooxygenase-2 inhibitors. Bioorg. Med. Chem. Lett. 2002; 12:1315-1318.
23. Harman CA, Turman MV, Kozak KR, Marnett LJ, Smith WL, 40. Garavito RM. Structural basis of enantioselective inhibition of cyclooxygenase-1 by S-alpha-substituted indomethacin ethanolamides. J. Biol. Chem. 2007; 282:28096-28105.41.
24. Vane JR, Flower RJ, Botting RM. History of aspirin and its mechanism of action Stroke 1990; 21(suppl IV):IV-12-23.
25. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biol. 1971; 231:232-235.
26. Roth GJ, Stanford N, Majerus PW. Acetylation of prostaglandin synthase by aspirin. Proc. Natl. Acad. Sci. U.S.A 1975; 72:42.3073-3076.
27. Van Der Ouderaa FJ, Buytenhek M, Nugteren DH, Van Dorp 43. DA. Acetylation of prostaglandin endoperoxide synthetase with acetylsalicylic acid, Eur. J. Biochem. 1980; 109:1-8.
28. Hochgesang GP, Marnett LJ.. Tyrosine 385 is critical for acetylation of cyclooxygenase-2 by aspirin. J. Am. Chem. Soc. 2000; 122: 6514-6515.44.
29. Loll PJ, Picot D, Garavito RM. The structural basis of as pirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nature Struct. Biol. 1995; 2:637-642.45.
30. Karthein R, Dietz R, Nastainczyk W, Ruf HH. Higher oxidation states of prostaglandin H synthase. EPR study of a transient tyrosyl radical in the enzyme during the peroxidase reaction. Eur. J. Biochem. 1988; 171:313-320.
31. Aronoff DM, Boutaud O, Marnett LJ, Oates JA. Inhibition of prostaglandin H2 synthases by salicylate is dependent on the oxidative state of the enzymes. J. Pharmacol. Exp. Ther. 2003; 304:589-595.
32. Boutaud O, Aronoff DM, Richardson JH, Marnett LJ, Oates JA. Determinants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H(2) synthases. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:7130-7135.
33. Capdevila JH, Morrow JD, Belosludtev YY, Beauchamp DR, DuBois RN, Falck Jr. The catalytic outcomes of the constitutive and the mitogen inducible isoforms of prostaglandin H2 synthase are markedly affected by glutathione and glutathione peroxidase(s). Biochemistry 1995; 34:3325-3337.
34. Kulmacz RJ, Wang LH. Comparison of hydroperoxide initiator requirements for the cyclooxygenase activities of prostaglandin H synthase-1 and -2. J. Biol. Chem. 1995; 270:24019-24023.
35. Mancini JA, O’Neill GP, Bayly C, Vickers PJ. Mutation of serine-516 in human prostaglandin G/H synthase-2 to methionine or aspirin acetylation of this residue stimulates 15-R-HETE synthesis. FEBS Lett. 1994; 342:33-37.
36. Rowlinson SW, Crews BC, Goodwin DC, Schneider C, Gierse JK, Marnett LJ. Spatial requirements for 15-HETE synthesis within the cyclooxygenase active site of murine COX-2: why acetylated COX-1 does not synthesize 15-(R)-HETE. J. Biol. Chem. 2000; 274:6586-6591.
37. Claria J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:9475-9479.
38. Kiefer JR, Pawlitz JL, Moreland KT, Stegeman RA, Hood WF, Gierse JK, Stevens AM, Goodwin DC, Rowlinson SW, Marnett LJ, Stallings WC, Kurumbail RG. Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature 2000; 405:97-101.
39. Rowlinson SW, Kiefer JR, Prusakiewicz JJ, Pawlitz JL, Kozak KR, Kalgutkar AS, Stallings WC, Kurumbail RG, Marnett LJ. A novel mechanism of cyclooxygenase-2 inhibition involving interactions with Ser-530 and Tyr-385. J. Biol. Chem. 2003; 278: 45763-45769.
40. Blobaum AL, Marnett LJ. Molecular determinants for the selective inhibition of cyclooxygenase-2 by lumiracoxib. J. Biol. Chem. 2007; 282:16379-16390.
41. Tannenbaum H, Berenbaum F, Reginster JY, Zacher J, Robinson J, Poor G, Bliddal H, Uebelhart D, Adami S, Navarro F, Lee A, Moore A, Gimona A. Lumiracoxib is effective in the treatment of osteoarthritis of the knee: a 13 week, randomised, double blind study versus placebo and celecoxib. Ann. Rheum. Dis. 2004; 63:1419-1426.
42. Lyseng-Williamson KA, Curran, Lumiracoxib MP. Drugs 2004; 64:2237-2248.
43. Clark K, Kulathila R, Koehn J, Rieffel S, Strauss A, Hu S, Kalfoglou M, Szeto D, Lasala D, Sabio M, Wang X, Marshall P. Crystal structure of the lumiracoxib:cyclooxygenase-2 complex. In: American Chemical Society: Book of Abstracts. 2004. pp. 22-26.
44. Moser P, Sallmann A, Wiesenberg I. Synthesis and quantitative structure-activity relationships of diclofenac analogues. J. Med. Chem. 1990; 33:2358-2368.
45. Blobaum AL, Marnett LJ. Structural and functional basis of cyclooxygenase inhibition. J. Med. Chem. 2007; 50:1425-1441.