THE LIVING WORLD
Unit Four. The Evolution and Diversity of Life
15. How We Name Living Things
15.5. How to Build a Family Tree
After naming and classifying some 1.5 million organisms, what have biologists learned? One very important advantage of being able to classify particular species of plants, animals, and other organisms is that we can identify species that are useful to humans as sources of food and medicine. For example, if you cannot tell the fungus Penicillium from Aspergillus, you have little chance of producing the antibiotic penicillin. In a thousand ways, just having names for organisms is of immense importance in our modern world.
Taxonomy also enables us to glimpse the evolutionary history of life on earth. The more similar two taxa are, the more closely related they are likely to be, for the same reason that you are more like your brothers and sisters than like strangers selected from a crowd. By looking at the differences and similarities between organisms, biologists can attempt to reconstruct the tree of life, inferring which organisms evolved from which other ones, in what order, and when. The evolutionary history of an organism and its relationship to other species is called phylogeny. The reconstruction and study of evolutionary trees, or phylogenetic trees, including the naming and classifying of organisms, is an area of study called systematics.
Cladistics
A simple and objective way to construct a phylogenetic tree is to focus on key characters that some organisms share because they have inherited them from a common ancestor. A clade is a group of organisms related by descent, and this approach to constructing a phylogeny is called cladistics. Cladistics infers phylogeny (that is, builds family trees) according to similarities derived from a common ancestor, so-called derived characters. Derived characters are defined as characters that are present in a group of organisms that arose from a common ancestor that lacked the character. The key to the approach is being able to identify morphological, physiological, or behavioral traits that differ among the organisms being studied and can be attributed to a common ancestor. By examining the distribution of these traits among the organisms, it is possible to construct a cladogram, a branching diagram that represents the phylogeny.
A cladogram of the vertebrates is shown in figure 15.5.
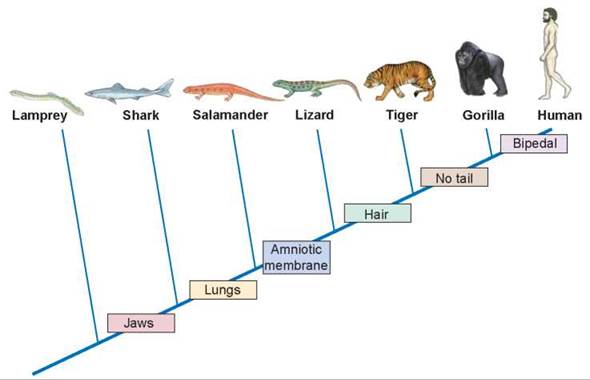
Figure 15.5. A cladogram of vertebrate animals.
The derived characters between the branch points are shared by all the animals to the right of each character and are not present in any organisms to the left of it.
Cladograms are not true family trees, derived directly from data that document ancestors and descendants like the fossil record does. Instead, cladograms convey comparative information about relative relationships. Organisms that are closer together on a cladogram simply share a more recent common ancestor than those that are farther apart. Because the analysis is comparative, it is necessary to have something to anchor the comparison to, some solid ground against which the comparisons can be made. To achieve this, each cladogram must contain an outgroup, a rather different organism (but not too different) to serve as a baseline for comparisons among the other organisms being evaluated, called the ingroup. For example, in figure 15.5 the lamprey is the outgroup to the clade of animals that have jaws. Comparisons are then made up the cladogram, beginning with lampreys and sharks, based on the emergence of derived characters. For example, the shark differs from the lamprey in that it has jaws, the derived character missing in the lamprey. The derived characters are in the colored boxes along the main line of the cladogram. Salamanders differ from sharks in that they have lungs, and so on up the cladogram.
Cladistics is a relatively new approach in biology and has become popular among students of evolution. This is because it does a very good job of portraying the order in which a series of evolutionary events have occurred. The great strength of a cladogram is that it can be completely objective. A computer fed the data will generate exactly the same cladogram time and again. In fact, most cladistic analyses involve many characters, and computers are required to make the comparisons. Although objective, the phylogenetic trees are not absolute. Phylogenetic trees are hypotheses, proposed explanations of how organisms may have evolved.
Sometimes cladograms are adjusted to “weight” characters, or take into account the variation in the “strength” (importance) of a character—the size or location of a fin, the effectiveness of a lung. For example, let’s say that the following are five unique events that occurred on September 11, 2001: (1) My cat was declawed, (2) I had a wisdom tooth pulled, (3) I sold my first car, (4) terrorists attacked the United States using commercial airplanes, and (5) I passed physics. Without weighting the events, each one is assigned equal importance. In a nonweighted cladistic sense, they are equal (all happened only once, and on that day), but in a practical, real-world sense, they certainly are not. One event, the terrorist attack, had a far greater impact and importance than the others. Because evolutionary success depends so critically on just such high-impact events, these weighted cladograms attempt to assign extra weight to the evolutionary significance of key characters.
Weighted cladograms are controversial. The problem with them is the systematist usually cannot always know how important each character is. The history of systematics has many examples of overemphasis or reliance on characters that later turned out to be less informative than had been thought. This is why many systematists now choose to weight all characters equally in cladograms.
Traditional Taxonomy
Weighting characters lies at the core of traditional taxonomy. In this approach, phylogenies are constructed based on a vast amount of information about the morphology and biology of the organism gathered over a long period of time. Traditional taxonomists use both ancestral and derived characters to construct their trees, whereas cladists use only derived characters. The large amount of information used by traditional taxonomists permits a knowledgeable weighting of characters according to their biological significance. In traditional taxonomy, the full observational power and judgment of the biologist is brought to bear—and also any biases he or she may have. For example, in classifying the terrestrial vertebrates, traditional taxonomists, shown by the phylogeny on the left in figure 15.6, place birds in their own class (Aves), giving great weight to the characters that made powered flight possible, such as feathers. However, a cladogram of vertebrate evolution, as shown on the right in figure 15.6, lumps birds in among the reptiles with crocodiles and dinosaurs. This accurately reflects their ancestry but ignores the immense evolutionary impact of a derived character such as feathers.
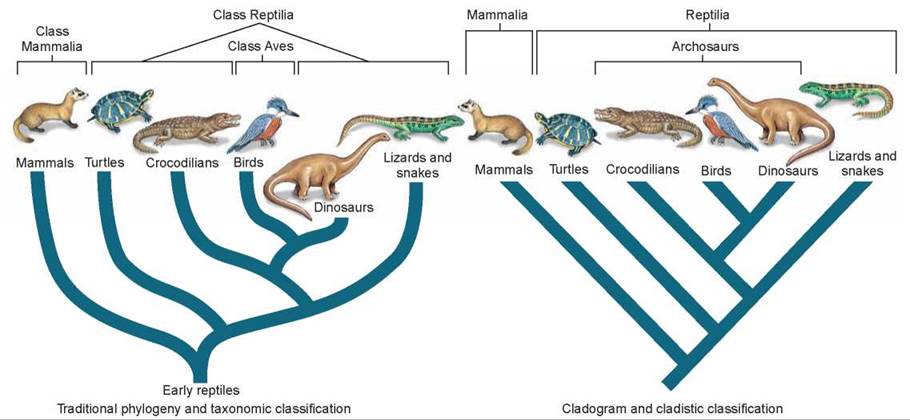
Figure 15.6. Two ways to classify terrestrial vertebrates.
Traditional taxonomic analyses place birds in their own class (Aves) because birds have evolved several unique adaptations that separate them from the reptiles. Cladistic analyses, however, place crocodiles, dinosaurs, and birds together (as archosaurs) because they share many derived characters, indicating a recent shared ancestry. In practice, most biologists adopt the traditional approach and consider birds as members of the class Aves rather than Reptilia.
Overall, phylogenetic trees based on traditional taxonomy are information-rich, while cladograms often do a better job of deciphering evolutionary histories. Traditional taxonomy is the better approach when a great deal of information is available to guide character weighting. For example, the cat family tree in figure 15.7 on the next page reflects a lot of knowledge about the different groups of felines. However, cladistics is the preferred approach when little information is available about how the character affects the life of the organism.
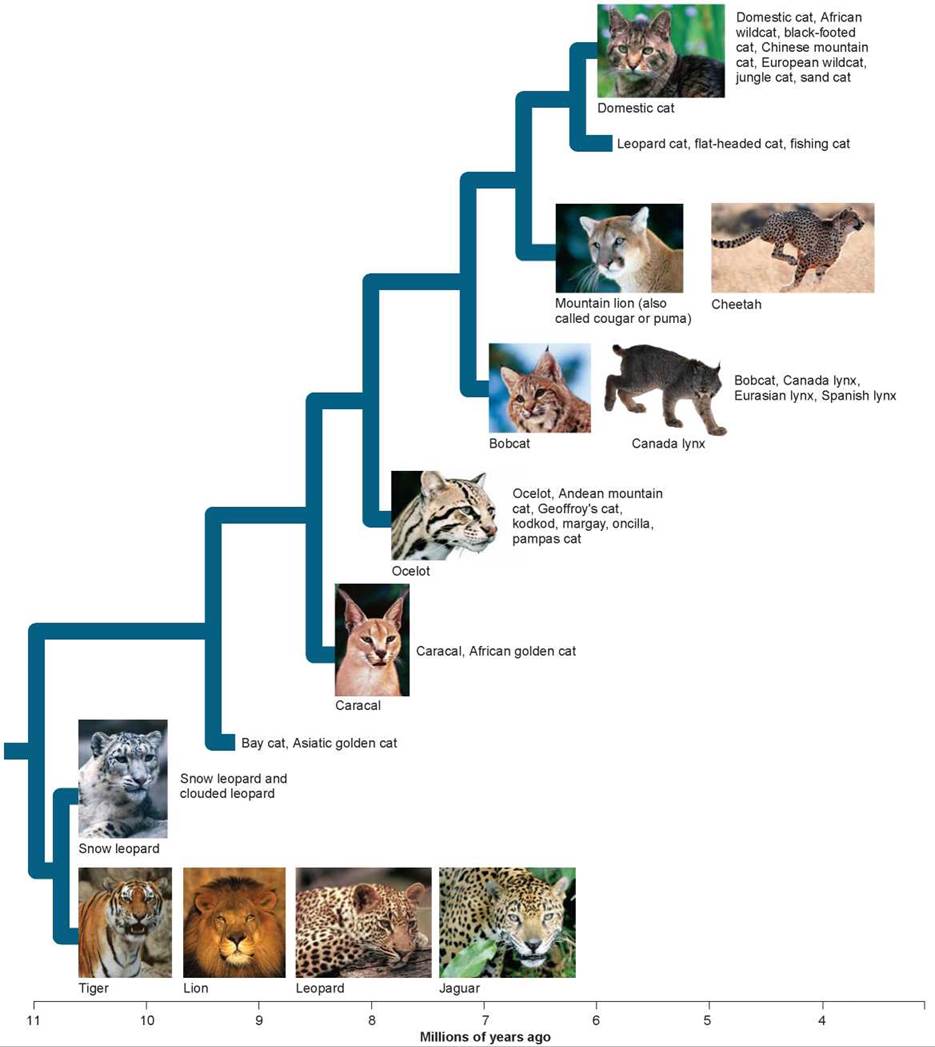
Figure 15.7. The cat family tree.
Recent studies of DNA similarities reported in 2006 have allowed biologists to construct this feline family tree of the eight major cat lineages and their individual species. Among the oldest of all cats are the four big panthers: tiger, lion, leopard, and jaguar. The other big cats, the cheetah and mountain lion, are members of a much younger lineage and are not close relatives of the big four. Domestic cats evolved most recently.
How Do You Read a Family Tree?
Evolutionary trees, more formally called phylogenies, have become an essential tool of modern biology, used to track the spread of mad cow disease, to trace an individual’s ancestry, and even to predict which horses might win the Kentucky Derby. Most importantly, evolutionary trees provide the main framework within which evidence for evolution is evaluated.
Given their central role in biology, it is important that you learn how to “read” a tree properly. Said simply, a phy- logeny or evolutionary tree is a depiction of lines of descent (figure 15.8). Its function is to communicate the evolutionary relationships among its elements. In a typical tree, individual genes, species, or other elements occupy the branch tips. Underneath, a network of branches connects to the base.

Figure 15.8. Tree as icon.
Darwin developed the metaphor of the "tree of life," living species tracing back through time to common ancestors in the same way that separate twigs on a tree trace back to the same branches.
The essential point in reading such a tree is to understand that the nodes (branching points) correspond to actual organisms that lived in the past. The tree does not illustrate the degree of similarity among the branch tips, but rather shows actual historical relationships. Although closely related organisms tend to be similar to one another, this is not the case if the rate of evolution is not uniform. As you saw in figure 15.6 previously, crocodiles are more closely related to birds than they are to lizards, even though anyone can see that crocodiles look a lot more like lizards than birds.
Once a tree is seen as a story, an historical account, it is easy to avoid confusion about relatives. The rule is simple: The more recently two species share a common ancestor, the more closely related they are. There is nothing new about this. This is how you refer to your relatives. You are more closely related to your first cousin than to your second cousin because your last common ancestor with your first cousin lived two generations ago (grandparents), while your last common ancestor with your second cousin lived three generations ago (great grandparents).
Now look how this works in an evolutionary tree depicting ancestry. Consider the tree diagram shown below. Some people erroneously conclude that a frog is more closely related to a shark than to a human. A frog is actually more closely related to a human than to a shark because the last common ancestor of a frog and a human (labeled x in the figure) is a descendant of the last common ancestor of a frog and a shark (labeled y in the figure), and thus lived more recently. It is that simple. Most problems with reading evolutionary trees come when one reads a tree along the tips. In the tree pictured below, this approach yields an orderly sequence from sharks to frogs to humans. This sequential way of reading a phylogeny is incorrect because it suggests a linear progression from primitive to advanced species, which in no way is justified by the tree. If so, the frog would be the ancestor of a living human.
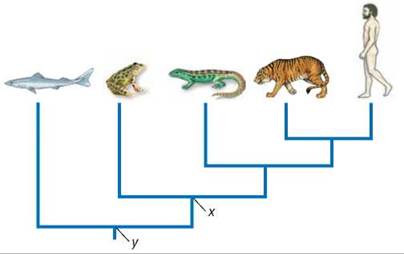
The correct way to read a tree is as a set of hierarchically nested groups, each of them a clade such as you encountered in figure 15.5. In the tree shown here, there are three meaningful clades: human-tiger, human-tiger-lizard, and human-tiger-lizard-frog.
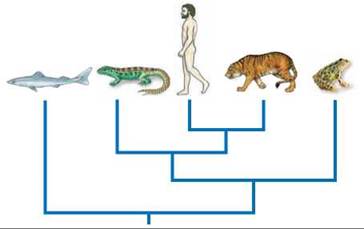
The difference between reading branch tips and reading clades becomes apparent if the branches are rotated so that the order of the tips is changed, as on the tree above. Although the order of branch tips is different, the branching patterns of descent—and the clade composition—is identical to the arrangement on the left. Evolutionary trees should be read by focusing on clade structure, which helps to emphasize that evolution is not a linear narrative.
Key Learning Outcome 15.5. An evolutionary tree depicts lines of descent, and is best read by focusing on clades. A cladogram is based on the order in which groups evolved, while a traditional taxonomic tree weights characters according to assumed importance.
Today’s Biology
DNA "Bar Codes"
The great diversity of life on earth is one of the glories of our planet. Wherever we look around us, we are surrounded by a profusion of life. A typical backyard contains hundreds of species of animals and plants, the same size slice of tropical rainforest contains orders of magnitude more. In North America alone, there are 709 identified species of birds, ranging in size from eagles with wingspans as long as your car to hummingbirds smaller than your thumb.
This profusion of species creates a problem that you might not at first anticipate. With so many species, how are you to know which is which? Animals and plants don't come with easy-to-read labels telling you to which species an individual belongs. Consider for example the two marsh wrens on the facing page. The darker individual on the right is from New Jersey, the lighter individual to its left from California. The difference in color between these two individuals tempts you to leap to an unwarranted conclusion, that the New Jersey specimen is an Eastern Marsh Wren—while the California specimen is a Western Marsh Wren. The conclusion is unwarranted because body color varies widely in both groups. There are lighter colored Eastern Marsh Wrens, and darker Western Marsh Wrens, leaving you in a quandary: Confronted with an individual marsh wren, how are you to determine which kind it is?
One solution to this dilemma is to take the specimen to a professional bird taxonomist, who will evaluate beak shape, plumage, and many other characters to correctly identify the bird. This is the approach Charles Darwin took in his study of the Galapagos. The finches, mockingbirds, and other birds he collected on the islands were studied and identified years later at the British Museum in London. However, there are not all that many taxonomists, and an awful lot of organisms whose identity we need to know.
Enter Dr. Paul Hebert of the University of Guelph in Ontario, Canada, with a deceptively simple suggestion, which is that organisms do in fact come with easy-to-read labels. Certain genes vary little among individuals of a species, but different species have different versions—why not let these genes serve as ID tags? DNA sequencing machines read off the order of nucleotides in batches of about 650 bases at a time, so he proposed examining the first 648 nucleotides of a gene called cytochrome c oxidase subunit 1 (CO1). Why this particular gene? For four reasons: First, because this gene is located on the mitochondrial DNA rather than on the nuclear chromosome, it is inherited solely from the mother and so escapes the shuffling of genetic material between generations that meiosis creates. Second, mitochondrial DNA is more stable than nuclear DNA, and can be obtained from museum specimens up to 20 years old. Third, in most animal species this gene has no inserted or deleted DNA, allowing all CO1 sequences to be lined up side-by-side for direct comparison.
Fourth, and most importantly, CO1 differences between individuals within a species are surprisingly small—just 2% of individuals differ at all along the 648-nucleotide stretch. This within-species uniformity is unusual—in a typical gene, many differences would be found between members of a species, so many as to overlap with individuals of closely related species. Not so for CO1. Perhaps because cytochrome oxidase plays such a critical role in oxidative metabolism, any changes within a species are rare, and when they do occur, spread rapidly through all members of the species. However, after two species have diverged, rare changes that occur in one species do not spread to the other, and so the two species accumulate defining differences.
How well does Hebert's suggestion work in practice? As a practical test, he set out to compare the first 648 nucleotides of the CO1 gene in mitochondrial DNA obtained from museum specimens of birds. He characterized 341 of the 709 known species of North American birds, and in every instance found a unique sequence characteristic of the species. While much larger samples will have to be examined to prove CO1 provides a distinctive signature, especially for closely related species, these initial results are very promising. You can see on the facing page the sequences Hebert found for the two marsh wrens we were discussing. Eastern and Western Marsh Wrens differ in 21 places, indicated by the lines between the two colored "bar codes.” Just as Hebert proposed, the CO1 sequences are easy-to-read labels that clearly identify an individual as being one kind of marsh wren, and not the other.
Dr. Hebert calls his approach "DNA bar coding” in analogy with the bar codes on supermarket items. The great potential of bar coding is that it solves the problem with which we introduced this essay, promising to allow anyone to correctly identify an unknown specimen in a direct and unambiguous fashion.
Of course, not every organism is a bird. How well does CO1 work as a bar code for other kinds of organisms? So far, it seems to work well for animals, but not as well for plants. Plant biologists have already begun to examine instead two genes found on chloroplast DNA, which seems better suited to distinguishing between plant species. While the central idea of bar coding is the use of a standard gene as a reference, the use of different genes for different major groups presents no major problem—few taxonomists would confuse a bird with a plant.
Bar coding is meeting a certain amount of resistance among taxonomists, who fear its widespread use will lead to sloppy science. The error that gives them pause is easily demonstrated with the marsh wren bar codes shown on the facing page: The 21 bar code differences serve to associate each individual with a "type” specimen that has a particular bar code. If that specimen is a different species, then so is the individual being tested. But the 21 differences do not establish by themselves that the two individuals are members of different species. Taxonomy cannot be reduced to a single gene, although identification of described species apparently can.

While DNA bar codes based on the CO1 gene appear to provide a useful ID tag for birds and potentially many other animal species, it is important to be very clear about the nature of the groups the bar codes identify. Any reproductively isolated line of descent can, and indeed would be expected to, develop a unique CO1 bar code. That does not mean the group is a separate species. For example, the aboriginal peoples of Australia have been reproductively isolated for many centuries, but if they were to have developed a unique CO1 bar code, that would not make them a separate species. It would imply no more and no less than genetic isolation.
This important distinction can be clearly seen in a recent study of Costa Rican butterflies. The skipper butterfly Astraptes fulgerator was first described in 1775 and ranges from Texas to Argentina. Over the course of 25 years of study, University of Pennsylvania ecologist Daniel Janzen has raised some 2,500 caterpillars of the Astraptes skipper that, as you can see in the photographs, come in 10 versions, each of which feeds on a different plant but all of which give rise to adults that look identical to one another and are all considered members of the same species.
Hearing of the bar code technology being developed by Hebert, Janzen removed one leg from each of his numerous preserved adult skipper butterflies and sent the samples to Dr. Hebert for analysis. Using the same CO1 bar code analysis that had proven so powerful in identifying bird species, Hebert found that Janzen's skipper collection fell neatly into 10 separate bar code clusters. All members of a cluster had nearly the same bar code, different from the other nine clusters. Importantly, the clusters matched the caterpillar groups! Each type of caterpillar had its own CO1 signature. The researchers concluded that in Costa Rica there is not one but 10 species of skipper, each with a strikingly different caterpillar. Janzen speculates that the groups diverged perhaps 4 million years ago, based on the amount of CO1 difference seen, each group specializing on a different caterpillar food plant. Is this a valid way to identify species? Some taxonomists are excited by the approach, others more cautious.
