Medical Microbiology
Section 5 Diagnosis and control
33 Attacking the enemy: antimicrobial agents and chemotherapy
Introduction
The interactions between host, microbial pathogen and antimicrobial agent can be considered as a triangle, and any alteration in one side will inevitably affect the other two sides (Fig. 33.1). In this chapter, two sides of the triangle will be examined in greater detail:
• the interactions between antimicrobial agents and microorganisms
• the interactions between antimicrobial agents and the human host.
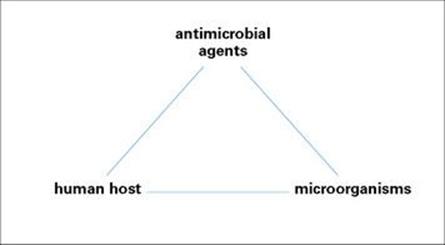
Figure 33.1 The interactions between antimicrobial agents, microorganisms and the human host can be viewed as a triangle. Any effect on one side of the triangle will have effects on the other two sides.
Laboratory aspects of antimicrobial susceptibility tests and assays will also be outlined. The third side of the triangle, the interactions between microorganisms and the human host, has been considered in detail in the preceding chapters. The concluding part of the present chapter will draw together the three sides of the triangle.
Selective toxicity
The term ‘selective toxicity’ was proposed by the immunochemist Paul Ehrlich (Box 33.1, Fig. 33.2). Selective toxicity is achieved by exploiting differences in the structure and metabolism of microorganisms and host cells; ideally, the antimicrobial agent should act at a target site present in the infecting organism, but absent from host cells. This is more likely to be achievable in microorganisms that are prokaryotes than in those that are eukaryotes, as the former are structurally more distinct from the host cells. (A comparison of the cellular organization of prokaryotic and eukaryotic cells is given in Ch. 1.) At the other end of the spectrum, viruses are difficult to attack because of their obligate intracellular lifestyle. A successful antiviral agent must be able to enter the host cell, but inhibit and damage only a virus-specific target. The desirable features of ideal antimicrobial agents are summarized in Box 33.2.
![]()
Box 33.1  Lessons in Microbiology
Lessons in Microbiology
Paul Ehrlich (1854–1915)
Just as Pasteur towers over immunomicrobiology, Ehrlich (Fig. 33.2) is the father figure of immunochemistry. His contributions to the science of medicine at all levels are quite extraordinary. He was the first to propose that foreign antigens were recognized by ‘side-chains’ on cells (1890), a brilliant insight that took 70 years to confirm. He also discovered the mast cell, invented the acid-fast stain for the tubercle bacillus, and devised a method to manufacture and commercialize a strong diphtheria antitoxin. He pioneered the development of antibiotics with his work on ‘606’ (or ‘Salvarsan’), a treatment for syphilis, for which he was denounced by the church for interfering with God’s punishment for sin.
While working on the treatment of infections caused by trypanosomes he set forth the concept of ‘selective toxicity’, as illustrated by the following quote: ‘But, gentlemen, it should be made clear that in general this task is much more complicated than that using serum therapy. These chemical agents, in contrast to the antibodies, may be harmful to the body. When such an agent is given to a sick organism, a difference must exist between the toxicity of this agent to the parasite and its toxicity to the host. We must always be aware of the fact that these agents are able to act on other parts of the body as well as on the parasites.’
Like Pasteur, he had a grasp of the continuum from the whole body to the cell and the three-dimensional structure of molecules, and throughout his life, he stressed the importance of molecular interaction as the basis of all biologic function; this is summed up in his famous maxim corpora non agunt nisi fixata or ‘things do not interact unless they make contact’. A Nobel Prize winner in 1908, his name was systematically eliminated from the records by the Nazi regime on account of his Jewish birth, but he was restored to honour by a reconstruction of his laboratory at the Seventh International Congress of Immunology in Berlin in 1989.
![]()

Figure 33.2 Paul Ehrlich (1854–1915).
![]()
Box 33.2 Desired Properties of a New Antimicrobial Agent
In the design of new antimicrobial agents, both antimicrobial activity and pharmacologic properties of the antibiotic for the host have to be considered.
Antimicrobial properties
• selectivity for microbial rather than mammalian targets
• cidal activity (antibacterial and antifungal agents)
• slow emergence of resistance
• narrow spectrum of activity.a
Pharmacologic activities
• non-toxic to the host
• long plasma half-life (once-a-day dosing)
• good tissue distribution including CSF
• low plasma-protein binding
• oral and parenteral dosing forms
• no interference with other drugs.
a The desired attribute depends on drug usage. Narrow-spectrum drugs cause less disturbance to normal flora and may contribute less to emergence of antibiotic resistance, whereas broad-spectrum compounds are more useful for empiric therapy and treatment of polymicrobial infections. CSF, cerebrospinal fluid.
![]()
Discovery and design of antimicrobial agents
The term ‘antibiotic’ has traditionally referred to natural metabolic products of fungi, actinomycetes and bacteria that kill or inhibit the growth of microorganisms. Antibiotic production has been particularly associated with soil microorganisms and, in the natural environment, is thought to provide a selective advantage for organisms in their competition for space and nutrients. Although the majority of antibacterial agents in clinical use today are derived from natural products of fermentation, most are then chemically modified (i.e. semi-synthetic) to improve their antibacterial or pharmacologic properties. However, some agents are totally synthetic (e.g. sulphonamides, quinolones). Therefore, the term ‘antibacterial’ or ‘antimicrobial’ agent is often used in preference to ‘antibiotic’. Agents used against fungi, parasites, and viruses can also be included under antimicrobials, but the terms antifungals, antiprotozoans, anthelmintics, and antivirals are more often used.
The discovery of new antimicrobial agents used to be entirely a matter of chance. Pharmaceutical companies undertook massive screening programmes searching for new soil microorganisms that produced antibiotic activity. In the light of our greater understanding of the mechanisms of action of existing antimicrobials, the processes have become rationalized, searching either for new natural products by target-site-directed screening or synthesizing molecules predicted to interact with a microbial target. Genomic approaches to the identification of novel targets have revolutionized this approach. In addition, knowledge of the crystal structure of the key enzymes involved in viral replication such as protease, reverse transcriptase and helicase leads to the design of new drugs. The steps in a rational design programme are summarized in Box 33.3.
![]()
Box 33.3 Rational Design of an Antimicrobial Agent
The discovery process of new antimicrobial agents has moved away from the random screening of soil microorganisms towards a rational design programme. From discovery to development and marketing can take up to 15 years and cost US$800 million. This list identifies different steps in this programme (average 10 years).
• Select an appropriate target.
• Identify a chemical lead (i.e. a new molecule with inhibitory activity on the target).
• Modify the lead compound to enhance potency.
• Evaluate in vitro activity.
• Evaluate in vivo activity and toxicity.
• Test in clinical trials and develop.
![]()
Classification of antibacterial agents
There are three ways of classifying antibacterial agents:
1. according to whether they are bactericidal or bacteriostatic
2. by target site
3. by chemical structure.
Some antibacterial agents are bactericidal, others are bacteriostatic
Some antibacterial agents kill bacteria (bactericidal), while others only inhibit their growth (bacteriostatic). Thus, the bactericidal process is irreversible, while bacteriostasis is reversible. Nevertheless, bacteriostatic agents are successful in the treatment of some infections because they prevent the bacterial population from increasing and host defence mechanisms can consequently cope with the static population. However, in immunocompromised patients, bacteriostatic drugs may be less efficacious, and certain infections (e.g., endocarditis) require a bactericidal drug even in an immunocompetent patient.
As a means of classification, the distinction between bactericidal and bacteriostatic agents is blurred because some agents are capable of killing some species, but are only bacteriostatic for others, e.g. chloramphenicol inhibits growth of Escherichia coli, but kills Haemophilus influenzae.
There are five main target sites for antibacterial action
A convenient way of classifying antibacterials is on the basis of their site of action. This classification does not allow an accurate prediction of which antibacterials will be active against which bacterial species, but it does help in the understanding of the molecular basis of antibacterial action, and conversely in the elucidation of many of the synthetic processes in bacterial cells. The five main target sites for antibacterial action are:
• cell wall synthesis
• protein synthesis
• nucleic acid synthesis
• metabolic pathways
• cell membrane function.
These targets differ to a greater or lesser degree from those in the host (human) cells and so allow inhibition of the bacterial cell without concomitant inhibition of the equivalent mammalian cell targets (selective toxicity).
Each target site encompasses a multitude of synthetic reactions (enzymes and substrates), each of which may be specifically inhibited by an antibacterial agent. A range of chemically diverse molecules may inhibit different reactions at the same target site (e.g. protein synthesis inhibitors).
Antibacterial agents have diverse chemical structures
Classification based on chemical structure alone is not of practical use, because there is such diversity. However, a combination of target site and chemical structure provides a useful working classification to organize antibacterial agents into specific families which will be discussed later in this chapter.
Resistance to antibacterial agents
Resistance to antibacterial agents is a matter of degree. In the medical setting, we define a resistant organism as one that will not be inhibited or killed by an antibacterial agent at concentrations of the drug achievable in the body after normal dosage. ‘Some men are born great, some achieve greatness, and some have greatness thrust upon them’ (William Shakespeare, Twelfth Night). Likewise, some bacteria are born resistant, others have resistance thrust upon them. In other words, some species are innately resistant to some families of antibiotics because they lack a susceptible target, are impermeable to or enzymatically inactivate the antibacterial agent. The Gram-negative rods with their outer membrane layer exterior to the cell wall peptidoglycan are less permeable to large molecules than Gram-positive cells. However, within species that are innately susceptible, there are also strains that develop or acquire resistance.
The genetics of resistance
In parallel with the rapid development of a wide range of antibacterial agents since the 1940s, bacteria have proved extremely adept at developing resistance to each new agent that comes along. This is illustrated for Staphylococcus aureus by the timeline shown in Figure 33.3. The rapidly increasing incidence of resistance associated with slowing down in the discovery of novel antibacterial agents to combat resistant strains is now recognized worldwide as a serious threat to the treatment of life-threatening infections.
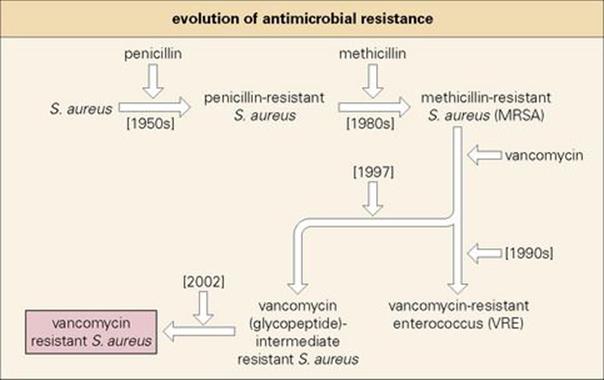
Figure 33.3 ‘Time line’ illustrating the chronological emergence of antibiotic resistance in Gram-positive cocci.
Chromosomal mutation may result in resistance to a class of antimicrobial agents (cross-resistance)
Resistance may arise from:
• a single chromosomal mutation in one bacterial cell resulting in the synthesis of an altered protein: for example, streptomycin resistance via alteration in a ribosomal protein, or the single amino acid change in the enzyme dihydropteroate synthetase resulting in a lowered affinity for sulphonamides. A mutational event could also alter (i.e., increase or decrease) the production of a protein resulting in increased resistance.
• a series of mutations, for example changes in penicillin-binding proteins (PBPs) in penicillin-resistant pneumococci.
In the presence of antibiotic, these spontaneous mutants have a selective advantage to survive and outgrow the susceptible population (Fig. 33.4A). They can also spread to other sites in the same patient or by cross-infection to other patients and therefore become disseminated. Chromosomal mutations are relatively rare events (i.e. usually found once in a population of 106–108 organisms) and generally provide resistance to a single class of antimicrobials (i.e. ‘cross-resistance’ to structurally related compounds).
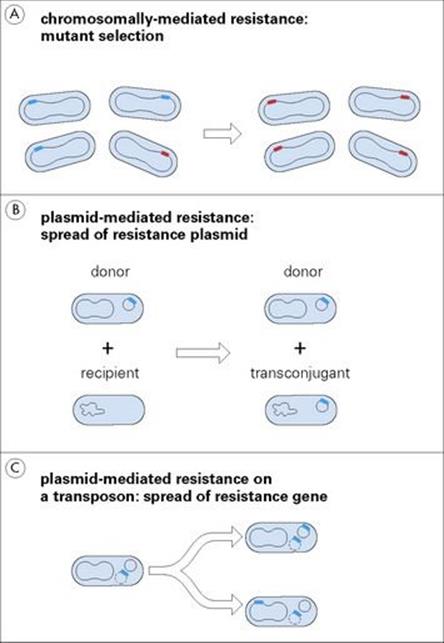
Figure 33.4 A chromosomal mutation (A) can produce a drug-resistant target, which confers resistance on the bacterial cell and allows it to multiply in the presence of antibiotic. Resistance genes carried on plasmids (B) can spread from one cell to another more rapidly than cells themselves divide and spread. Resistance genes on transposable elements (C) move between plasmids and the chromosome and from one plasmid to another, thereby allowing greater stability or greater dissemination of the resistance gene.
Genes on transmissible plasmids may result in resistance to different classes of antimicrobial agents (multiple resistance)
Not content with surviving the antibacterial onslaught by relying on random chromosomal mutation, bacteria are also able to acquire resistance genes on transmissible plasmids (Fig. 33.4B; see also Ch. 2). Such plasmids often code for resistance determinants to several unrelated families of antibacterial agents. Therefore a cell may acquire ‘multiple’ resistance to many different drugs (i.e. in different classes) at once, a process much more efficient than chromosomal mutation. This so-called ‘infectious resistance’ was first described by Japanese workers studying enteric bacteria, but is now recognized to be widespread throughout the bacterial world. Some plasmids are promiscuous, crossing species barriers, and the same resistance gene is therefore found in widely different species. For example, TEM-1, the most common plasmid-mediated beta-lactamase in Gram-negative bacteria, is widespread in E. coli and other enterobacteria and also accounts for penicillin resistance in Neisseria gonorrhoeae and ampicillin resistance in H. influenzae.
Resistance may be acquired from transposons and other mobile elements
Resistance genes may also occur on transposons; the so-called ‘jumping genes’, which by a replicative process are capable of generating copies which may integrate into the chromosome or into plasmids (see Ch. 2). The chromosome provides a more stable location for the genes, but they will be disseminated only as rapidly as the bacteria divide. Transposon copies moving from the chromosome to plasmids are disseminated more rapidly. Transposition can also occur between plasmids, for example, from a non-transmissible to a transmissible plasmid, again accelerating dissemination (Fig. 33.4C).
‘Cassettes’ of resistance genes may be organized into genetic elements called integrons
As discussed previously, antibiotic-resistance genes may individually reside on plasmids, the chromosome, or on transposons found in both locations. However, in some instances multiple resistance genes may come together in a structure known as an integron. As shown in Figure 33.5A, the integron encodes a site-specific recombination enzyme (int gene; integrase), which allows insertion (and also excision) of antibiotic-resistance gene ‘cassettes’ (resistance gene plus additional sequences including an ‘attachment’ region) into the integron attachment site (att). In classic operon fashion, a strong integron promoter controls transcription of the inserted genes. Based on their integration mechanism (integrase, etc.), integrons have been organized into different classes found in both Gram-negative and Gram-positive organisms. Whether acting as independent mobile genetic elements or inserted into transposons, integrons are capable of moving into a variety of DNA molecules, the overall hierarchy of which is depicted in Figure 33.5B. With their ability to capture, organize and rearrange different antibiotic-resistance genes, integrons represent an important mechanism for the spread of multiple antibiotic resistance in clinically important microorganisms.
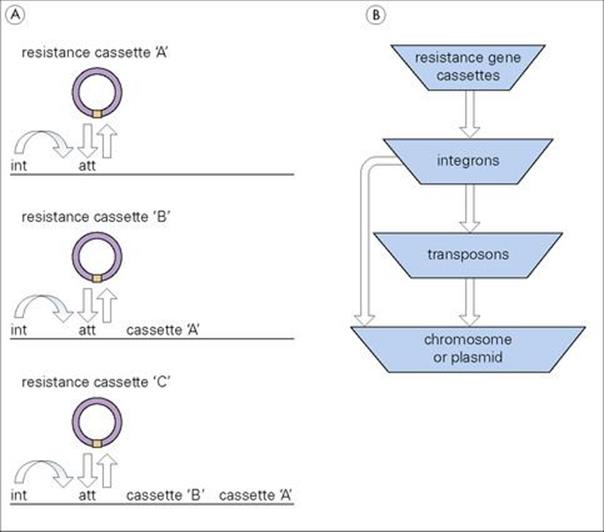
Figure 33.5 (A) Basic integron structure and (B) overall interrelationship between integrons and other DNA elements. att, integron attachment site; int, integrase.
Staphylococcal genes for methicillin resistance are organized into a unique cassette structure
Staphylococcal genes responsible for resistance to the antibiotic methicillin (discussed below) are found in a specialized cassette arrangement termed staphylococcal chromosomal casette mec (SCCmec). SCCmec inserts into a unique target site on the staphylococcal chromosome. The cassette represents a highly recombinogenic region which may not only rearrange internally but also serve as a target for the insertion of other resistance elements (e.g, transposons and plasmids).
Mechanisms of resistance
Resistance mechanisms can be broadly classified into three main types. These are summarized below, in Table 33.1 and described in more detail where relevant for each antibiotic in later parts of this chapter. Where bacterial mechanisms of antimicrobial resistance have been elucidated, they appear to involve the synthesis of new or altered proteins. As mentioned above, the genes encoding these proteins may be found on plasmids or the chromosome.
Table 33.1 Mechanisms of resistance can be classified into three main types
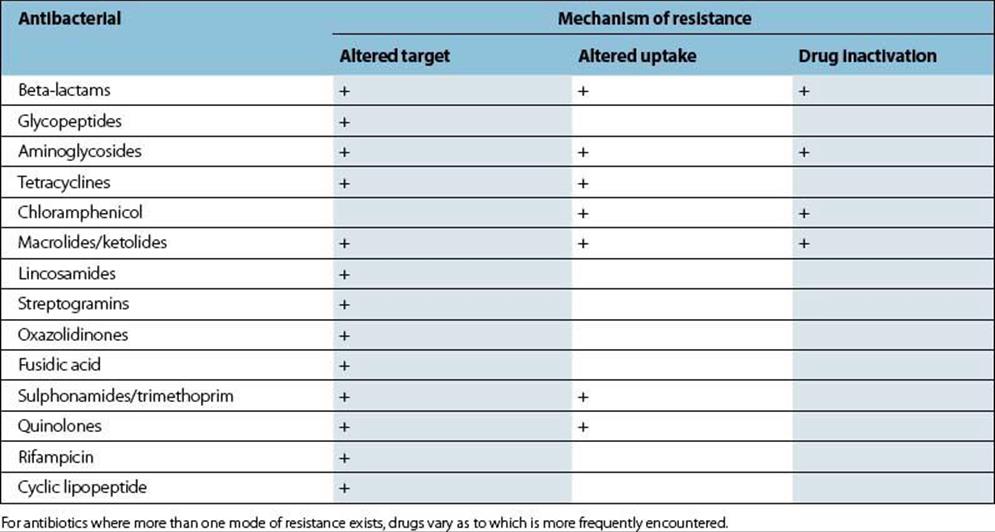
The target site may be altered
The target may be altered so that it has a lowered affinity for the antibacterial, but still functions adequately for normal metabolism to proceed. Alternatively, an additional target (e.g. enzyme) may be synthesized.
Access to the target site may be altered (altered uptake or increased exit)
This mechanism involves decreasing the amount of drug that reaches the target by either:
• altering entry, for example by decreasing the permeability of the cell wall
• pumping the drug out of the cell (known as an efflux mechanism).
Enzymes that modify or destroy the antibacterial agent may be produced (drug inactivation)
There are many examples of such enzymes, the most important being:
• beta-lactamases
• aminoglycoside-modifying enzymes
• chloramphenicol acetyl transferases.
These will be described in the relevant parts on these antibiotics.
Classes of antibacterial agents
The following parts of this chapter deal with groups of antibacterial agents based on their target site and chemical structure. In each case, the discussion attempts to summarize the answers to the questions set out in Table 33.2, reviewing the interactions between antibacterial agent and bacteria and between the antibacterial and the host (i.e. two sides of the triangle in Fig. 33.1).
Table 33.2 In order to understand the nature and optimum use of an antibacterial agent, the questions listed here must be answered
|
What is it? |
Chemical structure: natural or synthetic product |
|
What does it do? |
Target site, mechanism of action |
|
Where does it go? (and therefore preferred route of administration) |
Absorption, distribution, metabolism and excretion of the drug in the body of the host |
|
When is it used? |
Spectrum of activity and important clinical uses |
|
What are the limitations to its use? |
Toxicity to the human host; lack of toxicity, i.e. resistance of the bacteria |
|
How much does it cost? |
Great variation between agents but cost is a serious limitation on availability of some agents in resource-poor countries |
Inhibitors of cell wall synthesis
Peptidoglycan, a vital component of the bacterial cell wall (see Ch. 2), is a compound unique to bacteria and therefore provides an optimum target for selective toxicity. Synthesis of peptidoglycan precursors starts in the cytoplasm; wall subunits are then transported across the cytoplasmic membrane and finally inserted into the growing peptidoglycan molecule. Several different stages are therefore potential targets for inhibition (Fig. 33.6). The antibacterials that inhibit cell wall synthesis are varied in chemical structure. The most important of these agents are the beta-lactams, the largest group, and the glycopeptides which are active only against Gram-positive organisms. Bacitracin (primarily used topically) and cycloserine (mainly used as a ‘second-line’ medication for treatment of tuberculosis, discussed later in this chapter) have many fewer clinical applications.
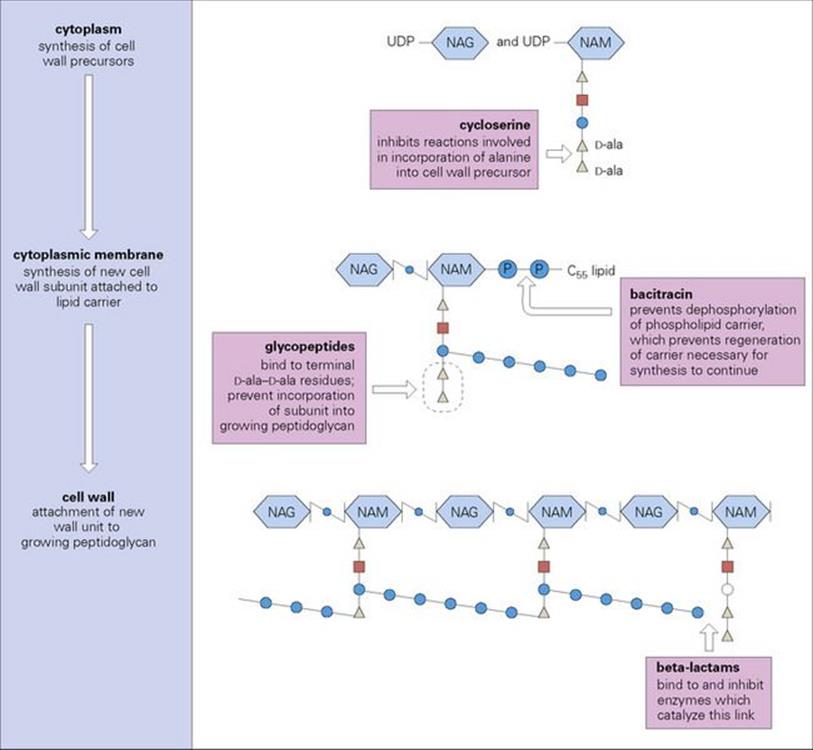
Figure 33.6 The synthesis of peptidoglycan is a complex process that begins in the cytoplasm, proceeds across the cytoplasmic membrane and leads to the attachment of new wall units to the growing peptidoglycan chain. This synthetic pathway can be inhibited at a variety of points by antibacterial agents. The precise mechanism of inhibition caused by glycopeptides such as vancomycin is unknown, but the mechanism of action of beta-lactams has now been fully elucidated (see text). NAG, N-acetyl glucosamine; NAM, N-acetyl muramic acid; UDP, uridine diphosphate.
Beta-lactams
Beta-lactams contain a beta-lactam ring and inhibit cell wall synthesis by binding to penicillin-binding proteins (PBPs)
Beta-lactams comprise a very large family of different groups of bactericidal compounds, all containing the beta-lactam ring. The different groups within the family are distinguished by the structure of the ring attached to the beta-lactam ring – in penicillins this is a five-membered ring, in cephalosporins a six-membered ring – and by the side chains attached to these rings (Fig. 33.7).
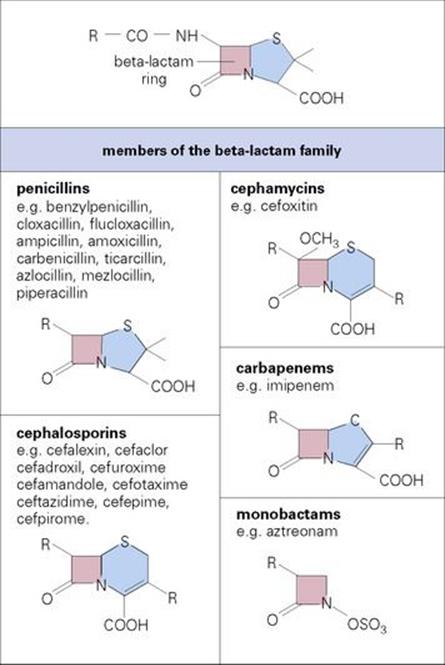
Figure 33.7 The beta-lactam family. The ring structure is common to all beta-lactams and must be intact for antibacterial action. Enzymes (beta-lactamases) that catalyse the hydrolysis of the beta-lactam bond render the agents inactive. The penicillins and cephalosporins are the major classes of beta-lactam antibiotics, but other members of the family, particularly the carbapenems and monobactams, are the focus of new developments.
PBPs are membrane proteins (e.g. carboxypeptidases, transglycosylases and transpeptidases) capable of binding to penicillin (hence the name PBP) and are responsible for the final stages of cross-linking of the bacterial cell wall structure. Inhibition of one or more of these essential enzymes results in an accumulation of precursor cell wall units, leading to activation of the cell’s autolytic system and cell lysis (Fig. 33.8).
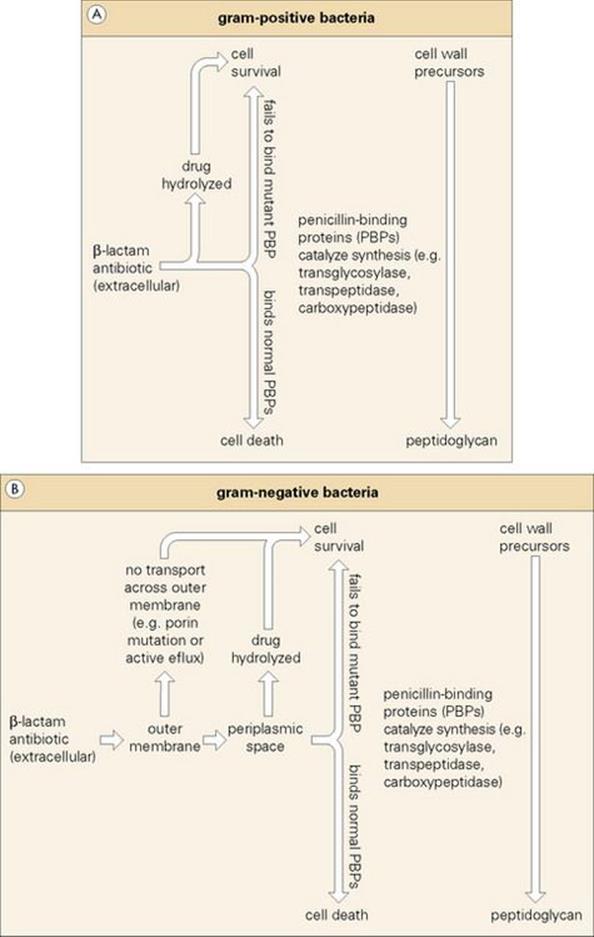
Figure 33.8 Penicillin-binding proteins (PBPs) play a key role in the final stages of peptidoglycan synthesis. They catalyse the cross-linkage of wall subunits, which are then incorporated into the cell wall. Beta-lactams are able to enter the cell (e.g. through pores in the outer membrane of Gram-negatives) and bind to the PBP. This prevents it from catalysing the cross-linkage of subunits, leading to their accumulation in the cell and the release of autolytic enzymes, which causes cell lysis. Within the periplasmic space of Gram-negatives (b1) beta-lactamases can inactivate beta-lactams before they reach their target PBPs, thereby protecting the cell from antibiotic action. Alternatively, mutant PBPs fail to bind beta-lactase, thus allowing peptidoglycan synthesis to occur. In Gram-positive bacteria (b2) beta-lactams may be extracellularly destroyed by beta-lactamases or rendered ineffective, as in Gram-negatives, by mutant PBPs.
Most beta-lactams have to be administered parenterally
Although the majority of beta-lactams have to be administered intramuscularly or intravenously, there are some orally active agents. Most achieve clinically useful concentrations in the cerebrospinal fluid (CSF) when the meninges are inflamed (as in meningitis) and the blood–brain barrier becomes more permeable. In general, they are not effective against intracellular organisms.
A few of the cephalosporins, notably cefotaxime, are metabolized to compounds with less microbiologic activity. All beta-lactams are excreted in the urine, and for some, such as benzylpenicillin, this is very rapid; hence the need for frequent doses. Probenecid can be administered concurrently to slow down excretion and maintain higher blood and tissue concentrations for a longer period of time.
Different beta-lactams have different clinical uses, but are not active against species that lack a cell wall
A vast array of beta-lactam antibiotics are currently registered for clinical use. Some, such as penicillin, are active mainly against Gram-positive organisms, whereas others (e.g. semi-synthetic penicillins, carboxypenems, monobactams, second-, third- and fourth-generation cephalosporins) have been developed for their activity against Gram-negative rods. Only the more recent beta-lactams are active against innately more resistant organisms such as Pseudomonas aeruginosa (Table 33.3).
Table 33.3 Characteristics of representative beta-lactams
|
Drug class |
Category |
General spectrum of activity |
|
Penicillins |
||
|
Penicillin G, Va |
Natural penicillin |
Gram-positive bacteria |
|
Cloxacillina |
|
|
|
Amoxicillina,b |
|
Gram-positive bacteria |
|
Carbenicillina |
Semisynthetic (carboxy) penicillin |
Gram-positive bacteria |
|
Cephalosporins |
||
|
Cefadroxila |
|
|
|
Cefaclora |
|
|
|
Cefdinira |
|
|
|
Cefepime |
|
Improved activity against Gram-negative bacteria |
|
Ceftaroline |
Anti-MRSA |
Improved activity, especially against MRSA |
|
Cephamycinc |
||
|
Cefmetazole |
Gram-positive bacteria |
|
|
Carbapenems |
||
|
Ertapenem |
Gram-positive and Gram-negative bacteria |
|
|
Monobactams |
||
|
Aztreonam |
Gram-negative bacteria including Haemophilus influenza and Pseudomonas aeruginosa |
 Gram-positive bacteria
Gram-positive bacteriaAlthough there are many beta-lactam agents available, the most commonly used ones are listed, together with their main indications.
a Oral formulation available.
b Can be formulated in combination with beta-lactamase inhibitors (see Fig. 33.9).
c Often classified with second generation cephalospoxins.
It is important to remember that beta-lactams are not active against species that lack a cell wall (e.g. Mycoplasma) or those with very impenetrable walls such as mycobacteria, or intracellular pathogens such as Brucella, Legionella and Chlamydia.
Resistance to beta-lactams may involve one or more of the three possible mechanisms
Resistance by alteration in target site
Methicillin-resistant staphylococci (e.g. Staph. aureus, Staph. epidermidis – MRSA, MRSE, respectively) synthesize an additional PBP (PBP2a) which has a much lower affinity for beta-lactams than the normal PBPs and is therefore able to continue cell wall synthesis when the other PBPs are inhibited. Although the mecA gene which codes for PBP2a is present on the chromosome in all cells of a resistant population, in many instances it may only be transcribed in a proportion of the cells, resulting in a phenomenon known as ‘heterogeneous resistance’. In the laboratory, special cultural conditions are used to enhance expression and demonstrate resistance. Methicillin-resistant staphylococci commonly produce beta-lactamase (see below) and are resistant to all other beta-lactams with the exception of ceftaroline, the first cephalosporin approved by the US FDA for activity against MRSA. This cephalosporin binds to PBP2a with an affinity 2000-fold better than other beta-lactams, and is thus effective in treating infections caused by MRSA.
Other organisms such as Streptococcus pneumoniae, Neisseria gonorrhoeae and Haemophilus influenzae may also utilize PBP changes to achieve beta-lactam resistance, which may vary depending on the compound employed.
Resistance by alteration in access to the target site
This mechanism is found in Gram-negative cells where beta-lactams gain access to their target PBPs by diffusion through protein channels (porins) in the outer membrane. Mutations in porin genes result in a decrease in permeability of the outer membrane and hence resistance. Strains resistant by this mechanism may exhibit cross-resistance to unrelated antibiotics that use the same porins.
Resistance by production of beta-lactamases
Beta-lactamases are enzymes that catalyse the hydrolysis of the beta-lactam ring to yield microbiologically inactive products. Genes encoding these enzymes are widespread in the bacterial kingdom and are found on the chromosome and on plasmids.
The beta-lactamases of Gram-positive bacteria are released into the extracellular environment (Fig. 33.8A) and resistance will only be manifest when a large population of cells is present. The beta-lactamases of Gram-negative cells, however, remain within the periplasm (Fig. 33.8B).
To date, hundreds of different beta-lactamase enzymes have been described. All have the same function but with differing amino acid sequences that influence their affinity for different beta-lactam substrates. Some enzymes specifically target penicillins or cephalosporins, while others are especially troublesome in broadly attacking most beta-lactam compounds (i.e. extended-spectrum beta-lactamases, ESBLs). Some beta-lactam antibiotics (e.g. carbapenems) are hydrolysed by very few enzymes (beta-lactamase stable), whereas others (e.g. ampicillin) are much more labile. Beta-lactamase inhibitors such as clavulanic acid (Fig. 33.9) are molecules that contain a beta-lactam ring and act as ‘suicide inhibitors’, binding to beta-lactamases and preventing them from destroying beta-lactams. They have little bactericidal activity of their own.
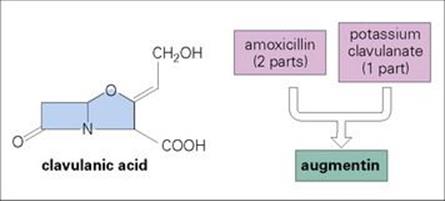
Figure 33.9 Clavulanic acid, a product of Streptomyces clavuligerus, inhibits the most common beta-lactamases (e.g. TEM enzymes) and allows amoxicillin to inhibit cells producing these enzymes. Augmentin is the most widely used of these combination drugs. Other combinations include ticarcillin and clavulanic acid, and piperacillin and tazobactam.
Side effects
Toxic effects of beta-lactam drugs include mild rashes and immediate hypersensitivity reactions
Statistics regarding allergy to beta-lactam drugs are complicated by the fact that the problem historically involves self-reporting by patients who are often mistaken in their ‘diagnosis’. Nevertheless, serious allergy to beta-lactam drugs in the form of an immediate (type 1) hypersensitivity reaction may occur in ca. 0.5–4% of patients, although anaphylaxis occurs much less frequently (ca. 0.004 to 0.04% of penicillin treatment courses). Mild idiopathic reactions, usually in the form of a rash, are more common (ca. 25% of treatment courses), especially with ampicillin. Patients who are allergic to penicillin are often allergic to cephalosporins (less with third-generation compounds) and vice versa, but aztreonam, a monobactam, shows negligible cross-reactivity.
Neurotoxicity and seizures can occur with all the beta-lactams if improperly dosed for body weight and kidney function, especially in patients with renal impairment. This toxicity is manifest as fits, unconsciousness, myoclonic spasms and hallucinations. Carbenicillin can cause platelet dysfunction and sodium overload (because it is given as a sodium salt), especially in patients with liver failure, renal failure and congestive heart failure.
Glycopeptides
Glycopeptides are large molecules and act at an earlier stage than beta-lactams
Glycopeptides include vancomycin and teicoplanin. Both are very large molecules and therefore have difficulty penetrating into Gram-negative cells. Teicoplanin is a natural complex of five different but closely related molecules.
Glycopeptides are bactericidal and interfere with cell wall synthesis by binding to terminal d-alanine-d-alanine at the end of pentapeptide chains that are part of the growing bacterial cell wall structure (see Fig. 33.6). This binding inhibits the transglycosylation reaction and prevents incorporation of new subunits into the growing cell wall. As glycopeptides act at an earlier stage than beta-lactams, it is not useful to combine glycopeptides and beta-lactams in the treatment of infections.
Vancomycin and teicoplanin must be given by injection for systemic infections
Vancomycin and teicoplanin are not absorbed from the gastrointestinal tract and do not penetrate the CSF in patients without meningitis. However, bactericidal concentrations are achieved in most patients with meningitis because of the increased permeability of the blood–brain barrier. Excretion is via the kidney.
Both vancomycin and teicoplanin are active only against Gram-positive organisms
Vancomycin and teicoplanin are used mainly for:
• the treatment of infections caused by Gram-positive cocci and Gram-positive rods that are resistant to beta-lactam drugs, particularly multiresistant Staphylococcus aureus and Staphylococcus epidermidis
• for patients allergic to beta-lactams
• the treatment of Clostridium difficile in antibiotic-associated colitis, although concerns that this may promote emergence of glycopeptide-resistant enterococci in the gut flora have led to the increasing use of alternative compounds.
Resistance
Some organisms are intrinsically resistant to glycopeptides
As mentioned previously, Gram-negative bacteria are ‘naturally’ resistant to the glycopeptides, since these compounds are too large to efficiently move through the outer membrane to the peptidoglycan. Other organisms have an altered glycopeptide target, such as pentapeptides, terminating in d-alanine-d-lactate (e.g. Erysiplothrix, Leuconostoc, Lactobacillus and Pediococcus) or d-alanine-d-serine (e.g. Enterococcus gallinarum, Enterococcus casseliflavus).
Organisms may acquire resistance to glycopeptides
Historically, the most clinically relevant acquired glycopeptide resistance has been observed in Enterococcus faecium and Enterococcus faecalis (vancomycin-resistant enterococci; VRE), first reported by investigators in the UK in 1986. Since that time, a variety of resistance phenotypes have been described which can be differentiated by transferability (e.g. plasmid association), inducibility and extent of resistance (Table 33.4). The genes associated with the highest levels of glycopeptide resistance are vanA, vanB, and vanD which encode a ligase producing pentapeptides terminating in d-alanine-d-lactate.
Table 33.4 Characteristics of glycopeptide resistance in enterococci
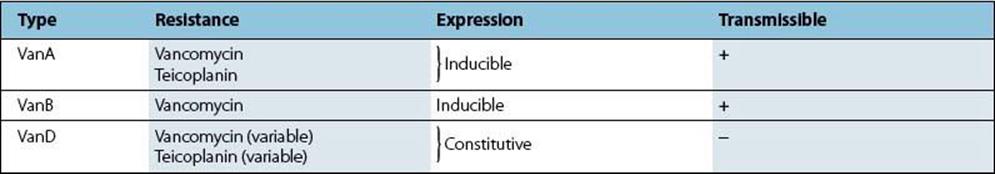
VanA is the best understood mechanism of acquired glycopeptide resistance
VanA-type glycopeptide resistance has been the most extensively studied and is characterized by inducible high-level resistance to both vancomycin and teicoplanin. VanA is associated with transposable elements related to Tn1546 (ca. 11 kb in size) which may be chromosomal or plasmid (transferable) in nature.
VanB is associated with inducible high-level resistance to vancomycin but not teicoplanin (although teicoplanin resistance can be induced by prior exposure to vancomycin). VanB resistance may be chromosomal or plasmid linked and is associated with very large transposable elements such as Tn1549 (34 kb).
VanD is chromosomal in nature and thus non-transferable, resulting in constitutive resistance to high levels of vancomycin but low levels of teicoplanin.
Glycopeptide resistance in the staphylococci occurs by mutation or by acquisition from the enterococci
Within the coagulase-negative staphylococci (CNS), Staphylococcus epidermidis and Staphylococcus haemolyticus are especially prone to development of glycopeptide resistance by mechanisms which remain incompletely understood. Nevertheless, resistant clinical and laboratory-generated isolates have been shown to differ from their susceptible counterparts in a variety of ways including changes in glycopeptide binding capacity, membrane proteins and cell wall synthesis and composition.
Coagulase-positive staphylococci (i.e. Staphylococcus aureus) showing decreased susceptibility to glycopeptides (but not fully resistant) were first described by Japanese investigators in 1996. The reduced susceptibility of these vancomycin-intermediate or glycopeptide-intermediate isolates (VISA or GISA, respectively) may be either homogeneously or heterogeneously expressed. In either case, ‘resistance’ is not associated with VanA, B, or D but, instead, appears to involve other mechanisms affecting cell wall composition (e.g. leading to increased thickness, etc.).
Unfortunately, high-level glycopeptide resistance has also been observed in Staph. aureus. This is due to the vanA gene (apparently acquired from VRE) residing on a staphylococcal plasmid.
Side effects
The glycopeptides are potentially ototoxic and nephrotoxic
Vancomycin is usually given by intravenous infusion, administered slowly to avoid ‘red-man’ syndrome due to histamine release. Particular care must be taken to prevent toxic concentrations accumulating in patients with renal impairment. Oral vancomycin is used for treatment of antibiotic-associated pseudomembranous colitis due to Clostridium difficile. Teicoplanin is less toxic than vancomycin and can be given by intravenous bolus and by intramuscular injection.
Inhibitors of protein synthesis
Although protein synthesis proceeds in an essentially similar manner in prokaryotic and eukaryotic cells, it is possible to exploit the differences (e.g. 70 S vs 80 S ribosome) to achieve selective toxicity. The process of translation of the messenger RNA (mRNA) chain into its corresponding peptide chain is complex, and a range of antibacterial agents act as inhibitors, although the full details of their mechanisms of action are not yet known (Fig. 33.10).
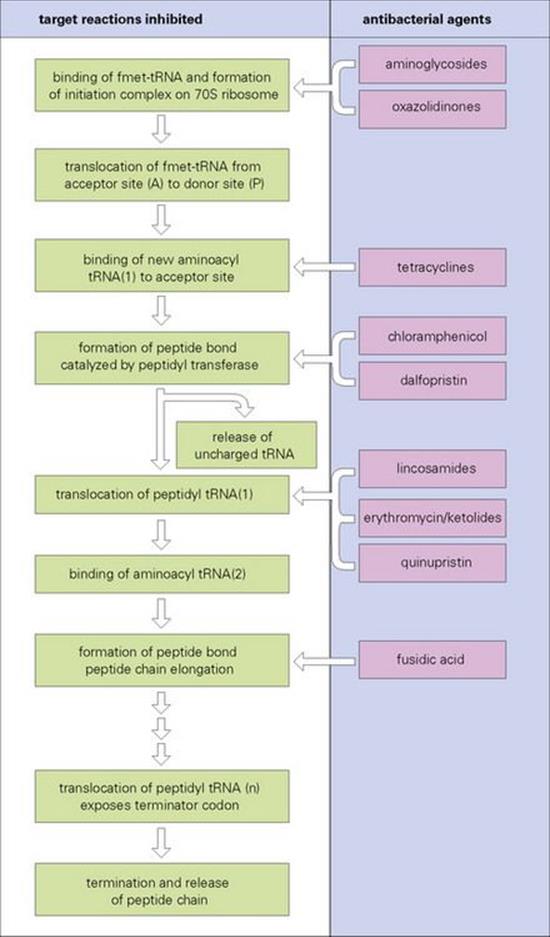
Figure 33.10 The synthetic pathway leading to the production of new protein in bacterial cells is extremely complex and still not fully elucidated. A number of different groups of antibacterial agents act by inhibiting proteins with specific reactions in this synthetic pathway. They can be grouped into those that act on the 30 S subunit of the ribosome (e.g. aminoglycosides and tetracyclines) and those that act on the 50 S subunit (e.g. chloramphenicol, lincosamides, erythromycin and fusidic acid). fmet-tRNA, formylmethionyl-transfer RNA.
Aminoglycosides
The aminoglycosides are a family of related molecules with bactericidal activity
The aminoglycosides contain either streptidine (streptomycin) or 2-deoxystreptamine (e.g. gentamicin; Table 33.5). The original structures have been modified chemically by changing the side chains to produce molecules such as amikacin and netilmicin that are active against organisms that have developed resistance to earlier aminoglycosides.
Table 33.5 Aminoglycoside-aminocyclitol antibiotics classified according to their chemical structure
|
4,6-distributed 2-deoxystreptamines |
|
|
Gentamicina |
Complex of 3 closely related structures; first aminoglycoside with broad spectrum |
|
Tobramycinb |
Activity very similar to gentamicin but slightly better against Pseudomonas aeruginosa |
|
Amikacin |
Semi-synthetic derivative of kanamycin; active against many gentamicin- resistant Gram-negative rods |
|
Netilmicinb |
Activity spectrum similar to amikacin: possibly lower toxicity |
|
4,5-disubstituted 2-deoxystreptamines |
|
|
Neomycinb |
Too toxic for parenteral use but has topical uses in decontaminating mucosal surfaces |
|
Streptidine-containing |
|
|
Streptomycinb |
Oldest aminoglycoside; now use restricted to treatment of tuberculosis |
They are also differentiated by the genus of microorganisms that produces them, and this is reflected in the spelling of the names.
a Micins from Micromonospora species.
b Mycins from Streptomyces species.
Aminoglycosides act by binding to specific proteins in the 30 S ribosomal subunit, where they interfere with the binding of formylmethionyl-transfer RNA (fmet-tRNA) to the ribosome (Fig. 33.10), thereby preventing the formation of initiation complexes from which protein synthesis proceeds. In addition, aminoglycosides cause misreading of mRNA codons and tend to break apart functional polysomes (protein synthesis by multiple ribosomes tandemly attached to a single mRNA molecule) into non-functional monosomes.
Aminoglycosides must be given intravenously or intramuscularly for systemic treatment
Aminoglycosides are not absorbed from the gut, do not penetrate well into tissues and bone, and do not cross the blood–brain barrier. Thus, they are usually administered as an intravenous infusion. Intrathecal administration of streptomycin is used in the treatment of tuberculous meningitis, and gentamicin may be administered by this route in the treatment of Gram-negative meningitis in neonates. Aminoglycosides are excreted via the kidney.
Gentamicin and the newer aminoglycosides are used to treat serious Gram-negative infections
Gentamicin, tobramycin, amikacin and netilmicin are important for the treatment of serious Gram-negative infections, including those caused by P. aeruginosa (Box 33.4). They are not active against streptococci or anaerobes, but are active against staphylococci. Against P. aeruginosa,amikacin is most active. Amikacin and netilmicin may be active against strains resistant to gentamicin and tobramycin (see below). Streptomycin is now reserved almost entirely for the treatment of mycobacterial infections. Neomycin is not used for systemic treatment, but can be used orally in gut decontamination regimens in neutropenic patients.
![]()
Box 33.4  Indications for Aminoglycoside Therapy
Indications for Aminoglycoside Therapy
Aminoglycosides are valuable additions to the clinician’s armamentarium despite their potential toxicity. They are important agents active against Gram-negative facultative bacteria and are often used in combination with beta-lactams to broaden the spectrum to include streptococci and some anaerobes, which are not susceptible to aminoglycosides alone. Resistance to aminoglycosides, particularly among enterobacteria and staphylococci, is mediated by the production of aminoglycoside-modifying enzymes, which react with groups on the aminoglycoside molecule to yield an altered aminoglycoside product. This competes with the unmodified aminoglycoside for uptake into the cell and binding to the ribosome.
Basic rule: use only in severe, life-threatening infections
• Gram-negative septicaemia (including Pseudomonas) usually in combination with beta-lactam
• Septicaemia of unknown aetiology arising from:a
• hospital-acquired infection
• malignancy
• immunosuppressive therapy
• major trauma, major surgery or major burns
• intravenous catheter
• urinary catheter
• extremes of age
• Bacterial endocarditis for synergy with beta-lactam
• Staphylococcus aureus septicaemia in combination with beta-lactam
• Pyelonephritis for difficult cases
• Post-surgical abdominal sepsis in combination with anti-anaerobe therapy.
a Every effort should be made to establish aetiology.
![]()
Production of aminoglycoside-modifying enzymes is the principal cause of resistance to aminoglycosides
Although relatively uncommon, resistance to aminoglycoside antibiotics may occur by alteration of the 30 S ribosomal target protein (e.g. a single amino acid change in the P12 protein prevents streptomycin binding). Resistance may also arise through alterations in cell wall permeability or in the energy-dependent transport across the cytoplasmic membrane.
Production of aminoglycoside-modifying enzymes is the most important mechanism of acquired resistance (Fig. 33.11). The genes for these enzymes are often plasmid-mediated, located on transposons, and transferable from one bacterial species to another. The enzymes alter the structure of the aminoglycoside molecule, thus inactivating the drug. The type of enzyme determines the spectrum of resistance of the organism containing it.
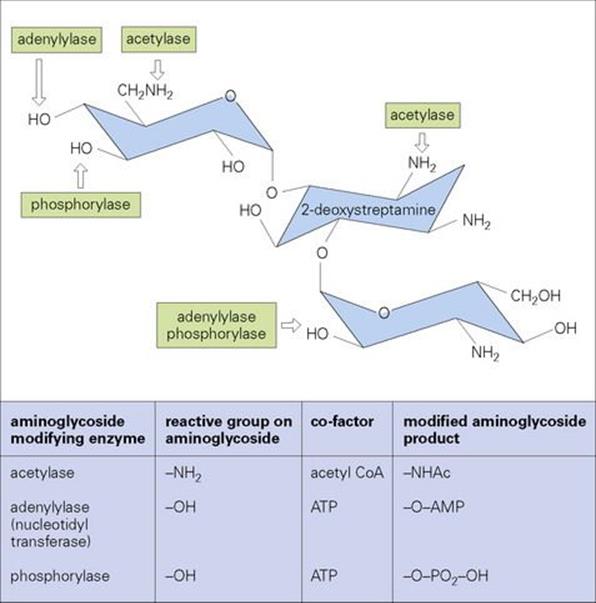
Figure 33.11 Prototype structure of aminoglycoside consisting of aminohexoses linked via glycosidic linkage to a central 2-deoxystreptamine nucleus. Hydroxyl and amino groups are sites at which these compounds can be inactivated by phosphorylation, adenylation or acetylation catalysed by enzymes produced by resistant strains.
The aminoglycosides are potentially nephrotoxic and ototoxic
The therapeutic ‘window’ between the serum concentration of aminoglycoside required for successful treatment and that which is toxic is small. Blood concentrations should be monitored regularly, particularly in patients with renal impairment. Netilmicin is reported to be one of the less toxic aminoglycoside antibiotics.
Tetracyclines
Tetracyclines are bacteriostatic compounds that differ mainly in their pharmacological properties rather than in their antibacterial spectra
Tetracyclines are a family of large cyclic structures that have several sites for possible chemical substitutions (Fig. 33.12).
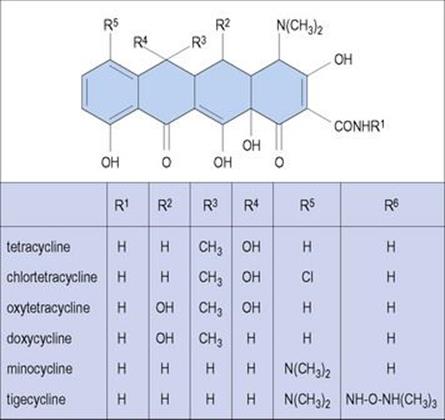
Figure 33.12 Tetracyclines are four-ring molecules with five different sites for substitution, thereby giving rise to a family of molecules with different substituents at different sites. Members of the family differ more in their pharmacologic properties than in their spectrum of activity.
Tetracyclines inhibit protein synthesis by binding to the small ribosomal subunit in a manner that prevents aminoacyl transfer RNA from entering the acceptor sites on the ribosome (see Fig. 33.10). While this process may occur with both prokaryotic and eukaryotic ribosomes, the selective action of tetracyclines is due to their much greater uptake by prokaryotic cells.
Tetracyclines are usually administered orally. Doxycycline and minocycline are more completely absorbed than tetracycline, oxytetracycline and chlortetracycline and so result in higher serum concentrations and less gastrointestinal upset because there is less inhibition of normal gut flora. Tetracyclines are well distributed and penetrate host cells to inhibit intracellular bacteria. They are excreted primarily in bile and urine.
Tetracyclines are active against a wide variety of bacteria, but their use is restricted due to widespread resistance
Tetracyclines are used in the treatment of infections caused by mycoplasmas, chlamydiae and rickettsiae. Resistance in other genera is common, due partly to the widespread use of these drugs in humans and also to their use as growth promoters in animal feed. The resistance genes are carried on a transposon, and new cytoplasmic membrane proteins are synthesized in the presence of tetracycline. As a result, tetracycline is positively pumped out of resistant cells (efflux mechanism). Although included with the tetracyclines (Fig. 33.12), tigecycline is a new member of a related class of compounds (glycylcyclines), derived from minocycline, with activity against bacteria resistant to tetracyclines.
Tetracyclines should be avoided in pregnancy and in children under 8 years of age
Tetracyclines suppress normal gut flora, resulting in gastrointestinal upset and diarrhea and encouraging overgrowth by resistant and undesirable bacteria (e.g. Staph. aureus) and fungi (e.g. Candida).
Interference with bone development and brown staining of teeth occurs in the fetus and in children. Systemic administration may cause liver damage. The potential for photosensitization is another caveat associated with the use of tetracyclines in all patients.
Chloramphenicol
Chloramphenicol contains a nitrobenzene nucleus and prevents peptide bond synthesis, with a bacteriostatic result
Chloramphenicol is a relatively simple molecule containing a nitrobenzene nucleus, which is responsible for some of the toxic problems associated with the drug (see below). Other derivatives have been produced, but none is in widespread clinical use.
Chloramphenicol has affinity for the large (50 S) ribosomal subunit where it blocks the action of peptidyl transferase, thereby preventing peptide bond synthesis (see Fig. 33.10). The drug has some inhibitory activity on human mitochondrial ribosomes (which are also 70 S) which may account for some of the dose-dependent toxicity to bone marrow (see below).
Chloramphenicol is well absorbed when given orally, but can be given intravenously if the patient cannot take drugs by mouth. Topical preparations are also available. It is well distributed in the body and penetrates host cells. Chloramphenicol is metabolized in the liver by conjugation with glucuronic acid to yield a microbiologically inactive form that is excreted by the kidneys.
Resistance and toxicity have limited the use of chloramphenicol
Chloramphenicol has been used in the treatment of bacterial meningitis (particularly H. influenzae) since the drug achieves satisfactory concentrations in the CSF. Chloramphenicol is active against a wide variety of bacterial species, both Gram-positive and Gram-negative, aerobes and anaerobes, including intracellular organisms. However, its potential serious toxic effects (see below) and issues of resistance have all but eliminated the systemic use of chloramphenicol in countries where alternative agents are readily available.
The most common mechanism of chloramphenicol resistance involves the inactivation of the drug by a plasmid-mediated enzymatic mechanism which is easily transferred within Gram-negative bacterial populations. Chloramphenicol acetyl transferases produced by resistant bacteria (Fig. 33.13) are intracellular, but are capable of inactivating all chloramphenicol in the immediate environment of the cell. Acetylated chloramphenicol fails to bind to the ribosomal target.
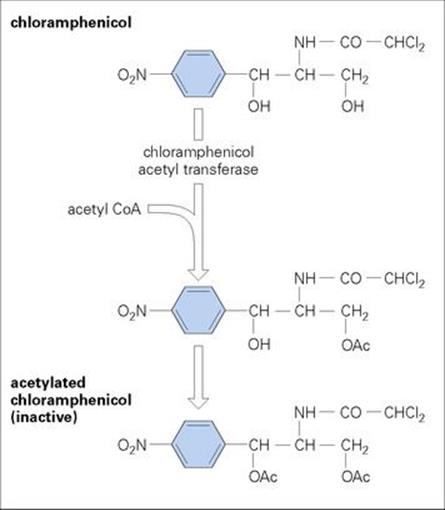
Figure 33.13 Resistance to chloramphenicol is mediated in some organisms by the production of a chloramphenicol acetyl transferase enzyme, which catalyses the addition of acetyl groups to the chloramphenicol molecule. This is a two-stage reaction producing acetylated chloramphenicol, which is inactive.
The most important toxic effects of chloramphenicol are in the bone marrow
Nitrobenzene is a bone marrow suppressant, and the structurally similar chloramphenicol molecule has similar effects. This toxicity takes two forms:
• dose-dependent bone marrow suppression, which occurs if the drug is given for long periods and is reversible when treatment is stopped
• an idiosyncratic reaction causing aplastic anaemia, which is not dose dependent and is irreversible. It can occur after treatment has stopped, but is fortunately very rare, occurring in about 1 in 30 000 patients treated.
Chloramphenicol is also toxic to neonates, particularly premature babies whose liver enzyme systems are incompletely developed. This can result in ‘grey baby syndrome’. Thus, chloramphenicol serum concentrations should be monitored in neonates.
Macrolides, lincosamides and streptogramins
These three groups of antibacterial agents share overlapping binding sites on ribosomes, and resistance to macrolides confers resistance to the other two groups. The clinically important drugs are the macrolide erythromycin, the lincosamide clindamycin, and the streptogramin combination quinupristin-dalfopristin.
Macrolides
Erythromycin is a widely used macrolide preventing the release of transfer RNA after peptide bond formation
The macrolides are a family of large cyclic molecules all containing a macrocyclic lactone ring (Fig. 33.14) and are bacteriostatic in activity. Erythromycin is the best known and most widely used, but some of the newer agents, such as azithromycin and clarithromycin, with improved activity and pharmacology may substitute erythromycin for specific indications. Spiramycin is another macrolide used in the prevention of congenital toxoplasmosis.
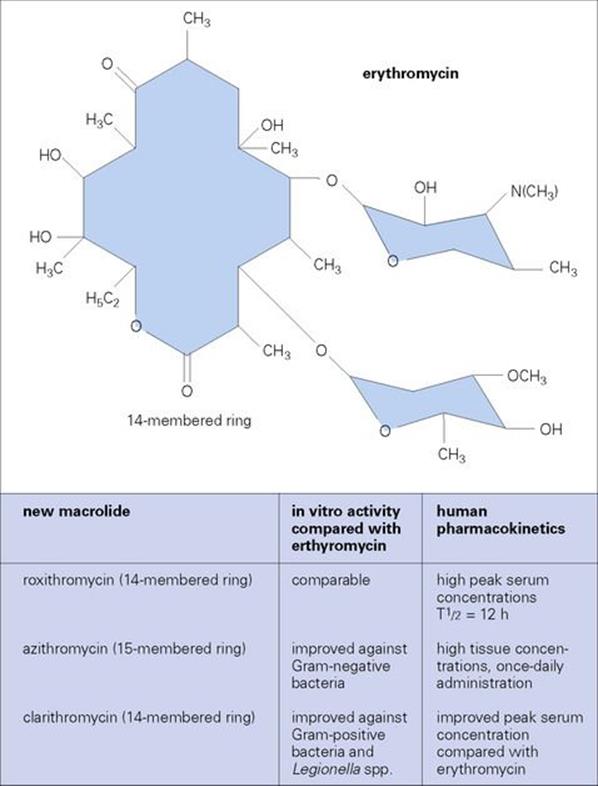
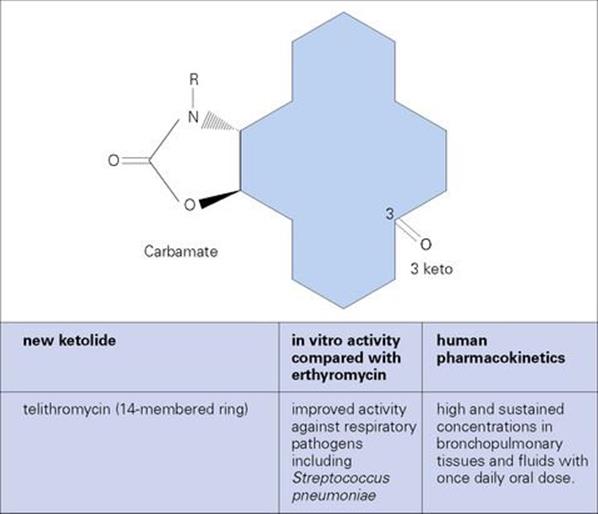
Figure 33.14 (A) The macrolides are antibacterial agents composed of large structures, which may be 14-, 15- or 16-membered rings. Erythromycin is the oldest and most widely used of these, but new agents with improved activity and fewer side effects are being developed. (B) Major differences in ketolide chemical structure compared to erythromycin (i.e. positions of 3-keto and carbamate on the ‘backbone’ ring structure).
Erythromycin binds to the 23 S ribosomal RNA (rRNA) in the 50 S subunit of the ribosome and blocks the translocation step in protein synthesis, thereby preventing the release of transfer RNA after peptide bond formation (see Fig. 33.10).
Erythromycin is usually administered by the oral route, but can also be given intravenously. It is well distributed in the body and penetrates mammalian cells to reach intracellular organisms. The drug is concentrated in the liver and excreted in the bile. A small proportion of the dose is recoverable in the urine.
Erythromycin is an alternative to penicillin for streptococcal infections, but resistant strains of streptococci are common
Erythromycin is active against Gram-positive cocci and is an important alternative treatment of infections caused by streptococci in patients allergic to penicillin. It is active against Legionella pneumophila and Campylobacter jejuni. It is also active against mycoplasmas, chlamydiae andrickettsiae and is therefore an important drug in the treatment of atypical pneumonia and chlamydial infections of the urinogenital tract.
Resistance is primarily due to either plasmid-encoded mef or erm genes, for efflux or alteration in the 23 S rRNA target by methylation of two adenine nucleotides in the RNA, respectively. The methylase enzyme may be either inducible or constitutively expressed. Erythromycin is a better inducer of resistance than the lincosamides, but strains resistant to erythromycin will also be resistant to lincomycin and clindamycin, so-called ‘MLS (macrolide-lincosamide-streptogramin) resistance’. Induction also varies between bacterial species, and resistant strains of Gram-positive cocci such as staphylococci and streptococci are common. In contrast to methylation, efflux is only active against macrolide drugs and does not confer lincosamide and streptogramin resistance.
Newer generation macrolides have fewer side effects than erythromycins
Erythromycin causes nausea and vomiting after oral administration in a significant number of patients. Jaundice is associated with some formulations of the drug.
Ketolides are new semi-synthetic derivatives of erythromycin with improved activity against respiratory pathogens
Modification of the macrolide ring structure (Fig. 33.14) provides ketolides with increased activity against a variety of Gram-positive (and some Gram-negative) bacteria, especially those associated with respiratory infections. Ketolides are administered orally and act in a manner similar to erythromycin. However, their higher affinity for the 50 S ribosomal subunit allows them to bind to ribosomes which are resistant to erythromycin. While active against methicillin-susceptible Staph. aureus that are either susceptible or inducibly resistant to erythromycin, ketolide activity is poor against erythromycin-resistant MRSA. In addition, telithromycin has had issues related to toxicity.
Lincosamides
Clindamycin inhibits peptide bond formation
Clindamycin is a chlorinated more active derivative of lincomycin and represents the most important drug in this class.
Lincosamides bind to the 50 S ribosomal subunit and inhibit protein synthesis in a manner similar to erythromycin (see Fig. 33.10), hence the MLS resistance combination noted above. The selectively toxic action results from a failure to bind to the equivalent mammalian ribosomal subunit.
Clindamycin is usually given orally, but can be administered intramuscularly or intravenously. It penetrates well into bone, but not into CSF, even when the meninges are inflamed. Clindamycin is actively transported into polymorphonuclear leukocytes and macrophages. It is metabolized in the liver to several products with variable antibacterial activity, and clindamycin activity persists in faeces for up to 5 days after a dose.
Clindamycin has a spectrum of activity similar to that of erythromycin
Clindamycin is much more active than erythromycin against anaerobes, both Gram-positive (e.g. Clostridium spp.) and Gram-negative (e.g. Bacteroides). However, Cl. difficile is resistant and may be selected in the gut, causing pseudomembranous colitis (see below). The activity of clindamycin against Staph. aureus and its penetration into bone make it a valuable drug in the treatment of osteomyelitis. Clindamycin is not active against aerobic Gram-negative bacteria because of poor penetration of the outer membrane.
As clindamycin is a less potent inducer of 23 S rRNA methylase (see MLS resistance above), erythromycin-resistant strains may appear susceptible to clindamycin in vitro. However, resistance will be manifest in vivo.
Pseudomembranous colitis caused by Cl. difficile was first noted following clindamycin treatment
Pseudomembranous colitis caused by Cl. difficile follows treatment with many antibiotics. The pathogenesis of this complication is described in Chapter 22, and it should be treated with metronidazole or oral vancomycin.
Streptogramins
The streptogramin formulation currently available is a mixture of streptogramin B and A compounds – quinupristin and dalfopristin, respectively (Fig. 33.15) – that are bacteriostatic individually but synergistically bactericidal in combination. Both compounds bind to 23 S RNA in the large (50 S) ribosomal subunit (dalfopristin facilitates binding of quinupristin). Dalfopristin inhibits protein synthesis at an earlier stage than quinupristin (see Fig. 33.10), and they together interfere with elongation and extension of peptide chains.
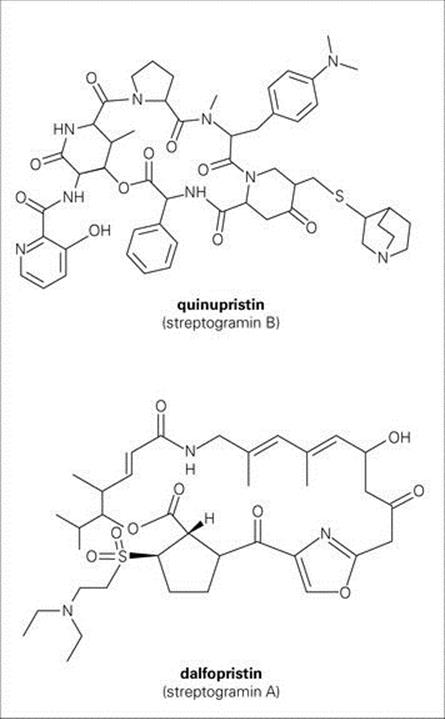
Figure 33.15 Chemical structure of the streptogramins.
Resistance is relatively uncommon but may develop by altering the quinupristin binding site (MLS resistance described above), enzymatic inactivation, or efflux.
The quinupristin–dalfopristin combination is active against Gram-positive cocci, including multidrug-resistant isolates. Activity is good against Enterococcus faecium but not E. faecalis (most probably due to an intrinsic efflux mechanism). However, there has been concern that commercial use of streptogramin compounds (e.g. virginiamycin) to prevent disease and promote growth in poultry could contribute to quinupristin–dalfopristin resistance among Gram-positive pathogens in humans.
Quinupristin–dalfopristin is administered intravenously and primarily metabolized in the liver.
Oxazolidinones
Oxazolidinones are a new class of synthetic bacteriostatic antimicrobial agents (Fig. 33.16). Linezolid, the oxazolidinone currently available, is active against a wide range of Gram-positive bacteria, including multiresistant strains. Linezolid inhibits initiation of protein synthesis (see Fig. 33.10) by targeting 23 S ribosomal RNA in the 50 S subunit in a manner which prevents formation of a functional 70 S complex.
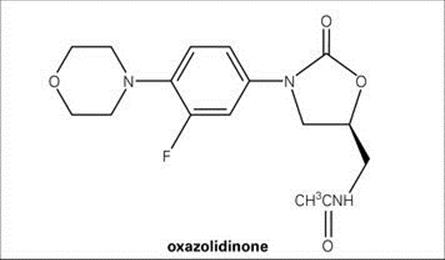
Figure 33.16 Chemical structure of oxazolidinones.
Due to the drug’s unique mechanism of action, resistance mutations (i.e. altered target) are rare and seen primarily in Enterococcus faecium.
Linezolid is administered orally or intravenously and is metabolized in the liver.
Fusidic acid
Fusidic acid is a steroid-like compound that inhibits protein synthesis
Fusidic acid is a bacteriostatic agent that inhibits protein synthesis by forming a stable complex with elongation factor EF-G (the bacterial equivalent of the human EF-2), guanosine diphosphate and the ribosome.
Fusidic acid can be administered orally or intravenously. It is well absorbed and penetrates well into tissues and bone, but not into the CSF. Topical preparations are also available, but their use should not be encouraged, because of the rapid emergence of resistance (see below). Fusidic acid is metabolized in the liver and excreted in the bile.
Fusidic acid is a treatment for staphylococcal infections, but should be used with other antistaphylococcal drugs to prevent emergence of resistance
Fusidic acid is active against Gram-positive cocci, and its most important use is in the treatment of staphylococcal infections resistant to beta-lactams or in patients who are allergic to alternative staphylococcal agents. Fusidic acid should be given in combination with another antistaphylococcal agent to prevent the emergence of resistant mutants with altered EF-G, which emerge rapidly in staphylococcal populations exposed to the drug.
Fusidic acid has few side effects
Occasionally, fusidic acid causes jaundice and gastrointestinal upset.
Inhibitors of nucleic acid synthesis
Antibacterial agents that act as inhibitors of nucleic acid synthesis do so in one of three main ways, as listed in Box 33.5.
![]()
Box 33.5 Inhibition of Nucleic Acid Takes Place at Different Stages in its Synthesis and Function, and Different Groups of Antimicrobial Agents are Involved
Inhibitors of DNA replication
• Quinolones
Inhibitors of RNA polymerase
• Rifampicin
Antimetabolites inhibiting precursor synthesis
• Sulphonamides, trimethoprim.
![]()
Quinolones
Quinolones are synthetic agents that interfere with replication of the bacterial chromosome
Quinolones represent a large family of bactericidal synthetic agents which, in a manner similar to the cephalosporins, can be generally grouped in categories or ‘generations’ based on their spectrum of activity (Table 33.6). Nalidixic acid is the first-generation prototype, but the addition of fluorine at position 6 of the main quinolone ring (i.e. fluoroquinolones) (Fig. 33.17) has improved antibacterial activity, leading to the synthesis of many additional, more commonly used, compounds.
Table 33.6 Characteristics of representative quinolones
|
Drug |
Categorya |
General spectrum of activity |
|
Nalidixic acid |
First generation |
Gram-negative bacteria (excluding Pseudomonas) |
|
Ciprofloxacin |
Second generation |
First-generation coverage but including Pseudomonas spp., and some Gram-positives (Staphylococcus aureus but not Streptococcus pneumoniae) |
|
Levofloxacin |
Third generation |
Second-generation coverage but improved Gram-positive coverage (penicillin-sensitive and resistant Strep. pneumoniae) and some activity against anaerobes |
|
Trovafloxacin |
Fourth generationb |
Third-generation coverage, expanded activity against anaerobes |
The most commonly used agents are listed, together with their main indications.
a All but first-generation compounds are fluoroquinolones.
b Associated with cases of acute liver failure; use reserved for life-threatening situations.
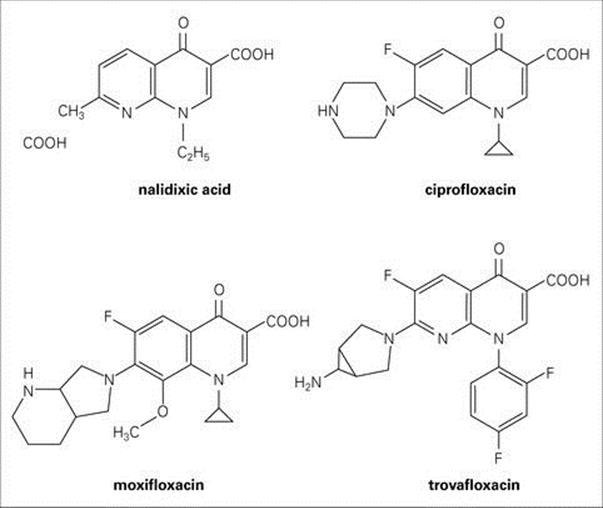
Figure 33.17 The quinolones form a large group of synthetic antibacterial agents.
The antibacterial activity of quinolones is due to their ability to inhibit the activity of bacterial DNA gyrase and topoisomerases. During replication of the bacterial chromosome, DNA gyrase produces and removes supercoils in DNA ahead of the replication fork to maintain the proper ‘tension’ required for efficient DNA duplication. Topoisomerase IV similarly acts to remove supercoils and to separate newly formed DNA ‘daughter’ strands after replication (Fig. 33.18). These enzymes thus act in concert to insure that the DNA molecule has the proper conformation for efficient replication and packaging within the cell. Quinolones are able to interfere with these essential enzymes in bacteria while not affecting their counterparts in mammalian cells.
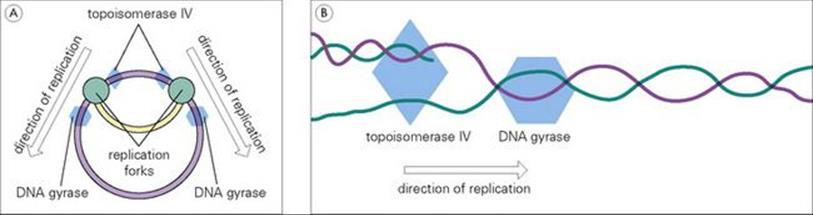
Figure 33.18 (A) An overview and (B) an enlarged view of the role played by bacterial gyrase and topoisomerase enzymes in replication of the bacterial chromosome.
Resistance to quinolones is usually chromosomally mediated
Chromosomally mediated resistance is exhibited in two forms:
• mutations, which change the target enzymes in a manner that affects quinolone binding
• changes in cell wall permeability, resulting in decreased uptake, or by efflux. These mechanisms may also lead to cross-resistance to other unrelated agents affected by the same process.
Plasmid-encoded quinolone resistance involves production of a protein (termed qnr) that protects the target DNA from quinolone binding. This protein has been shown to act in concert with a plasmid encoded enzyme capable of reducing the activity of some fluoroquinolones, resulting in increased levels of quinolone resistance.
Because of their safety and tolerability, quinolones are commonly used as alternatives to beta-lactam antibiotics for treating a variety of infections
Quinolones are primarily administered orally since they are readily absorbed from the gastrointestinal tract, achieving significant serum concentrations and good distribution throughout the body compartments. Excretion is mostly in the urine; however, drugs such as gatifloxacin and moxifloxacin are excreted to a significant amount in faeces.
Nalidixic acid does not achieve antibacterial systemic concentrations. It is only active against enterobacteria, and, although occasionally employed in treatment of urinary tract infections (see Ch. 20), its use has largely been replaced by the newer fluorinated compounds.
The newer quinolones have improved activity against Gram-negative rods, including P. aeruginosa. In addition to the treatment of urinary tract infections, the newer quinolones are useful for systemic Gram-negative infections and in the treatment of chlamydial and rickettsial infections. They are also useful in infections caused by other intracellular organisms, such as L. pneumophila and S. typhi, and in combination with other agents for ‘atypical’ mycobacteria. They have activity against staphylococci, but overall have only limited use against streptococci and enterococci (see Table 33.6).
Fluoroquinolones are not recommended for children or pregnant or lactating women because of possible toxic effects on cartilage development
Gastrointestinal disturbances are the most common side effect of quinolones. Neurotoxicity and photosensitivity reactions are less common. However, a notable exception is the potential for liver toxicity associated with trovafloxacin (see Table 33.6) which has prompted severe restrictions on its use. Gemifloxacin is associated with development of a rash which in some cases may be serious enough to require steroid treatment. This is especially a problem of women of child-bearing years or post-menopausal on hormone therapy. Gatifloxacin use has been impacted by issues of glucose homeostasis, sometimes severe enough to produce a coma. All fluoroquinolones have the potential to cause tendon ruptures in active patients who may tend to push their workout regimens. This risk is increased when quinolones and corticosteroids are simultaneously administered.
Rifamycins
Rifampicin is clinically the most important rifamycin and blocks the synthesis of mRNA
Rifampicin is the most important member of the rifamycin family in clinical use. It is a large molecule with a complex structure. Other family members such as rifabutin and rifapentine are also available. All are bactericidal in activity.
Rifampicin binds to DNA-dependent RNA polymerase and blocks the synthesis of mRNA. Selective toxicity is based on the far greater affinity for bacterial polymerases than for the equivalent human enzymes.
Rifampicin is administered orally, is well absorbed and is very well distributed in the body. It crosses the blood–brain barrier and reaches high concentrations in saliva. It also appears to have an affinity for plastics, which can be valuable in the treatment of infections involving prostheses.
Rifampicin is metabolized in the liver and excreted in bile. The compound is red, and urine, sweat and saliva of treated patients turns orange. This is harmless, although disturbing for the patient, but is good evidence of patient compliance.
The newer rifamycins, rifabutin and rifapentine are excreted more slowly than rifampicin, thereby allowing less frequent administration – a feature particularly attractive in the treatment of tuberculosis.
The primary use for rifampicin is in the treatment of mycobacterial infections, but resistance is a concern
While used primarily against mycobacteria, rifampicin may also be used for the prophylaxis of close contacts of meningococcal and Haemophilus meningitis. However, highly resistant meningococcal strains may emerge; thus short courses only (maximum 48 h) should be given (see Ch. 24).
While staphylococci rapidly develop resistance to rifampicin, the drug can be efficacious if used in combination with another agent, particularly in the treatment of prosthetic valve endocarditis (see Ch. 29).
Resistance is provided by chromosomal mutations that alter the RNA polymerase target, which then has lowered affinity for rifampicin and escapes inhibition. The prevalence of rifampicin-resistant M. tuberculosis is increasing, threatening the future of its use in antituberculosis therapy.
Rashes and jaundice are side effects of rifampicin treatment
Intermittent rifampicin can lead to hypersensitivity reactions.
Antimetabolites affecting nucleic acid synthesis
Several commonly used antimicrobial agents inhibit bacterial metabolic pathways including those which produce precursors for nucleic acid synthesis.
Sulphonamides
Sulphonamides are structural analogues of and act in competition with para-aminobenzoic acid
This group of molecules is produced entirely by chemical synthesis (i.e. they are not natural products). In 1935, the parent compound sulphanilamide became the first clinically effective antibacterial agent. The p-amino group is essential for activity, but modifications to the sulphonic acid side chain have produced many related agents (Fig. 33.19).
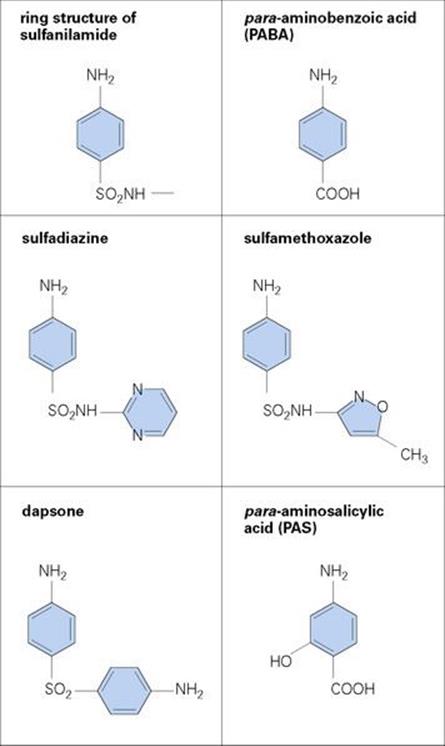
Figure 33.19 The ring structure of the sulphonamides is very similar to the structure of the normal substrate (PABA) of the dihydropteroate synthetase enzyme, which the sulphonamides inhibit. There are many different sulphonamides available and they differ in their pharmacologic properties more than in their spectrum of activity. Relatively few are now in common clinical use. Dapsone is important in the treatment of Mycobacterium leprae, and para-aminosalicylic acid is used for the treatment of M. tuberculosis.
Sulphonamides are bacteriostatic compounds that act in competition with para-aminobenzoic acid, PABA, for the active site of dihydropteroate synthetase, an enzyme that catalyses an essential reaction in the synthetic pathway of tetrahydrofolic acid (THFA), which is required for the synthesis of purines and pyrimidines and therefore for nucleic acid synthesis (Fig. 33.20). Selective toxicity depends on the fact that many bacteria synthesize THFA, whereas human cells lack this capacity and depend on an exogenous supply of folic acid. Bacteria that can use preformed folic acid are similarly unaffected by sulphonamides.
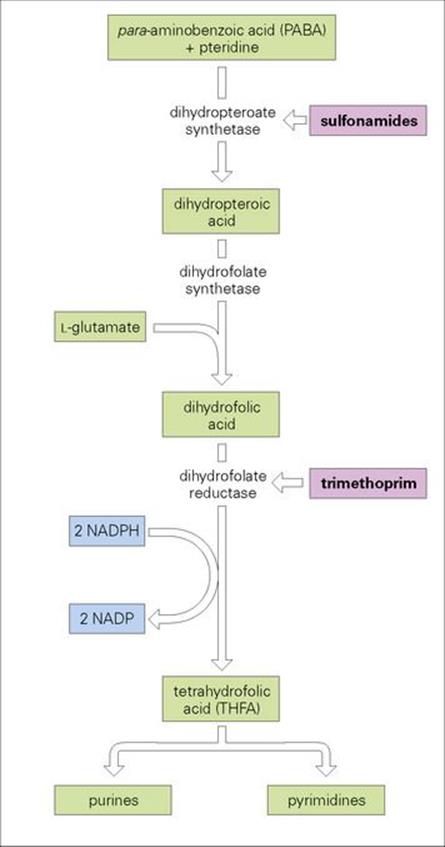
Figure 33.20 Sulphonamides and trimethoprim inhibit in series the steps in the synthesis of tetrahydrofolic acid by interacting with key enzymes in the pathway.
Sulphonamides are usually administered orally, often in combination with trimethoprim as co-trimoxazole (see below). Different molecules within the family differ in their solubility and penetrability. Metabolism occurs in the liver, and free and metabolized drug are excreted by the kidneys.
Sulphonamides are useful in the treatment of urinary tract infection, but resistance is widespread
The sulphonamides have a spectrum of activity primarily against Gram-negative organisms (except Pseudomonas). They are therefore useful in the treatment of urinary tract infections (see Ch. 20). However, susceptibility cannot be assumed, as resistance is widespread with plasmid-mediated genes coding for an altered dihydropteroate synthetase. This is essentially unchanged in its affinity for PABA, but has a greatly decreased affinity for the sulphonamide. A resistant cell therefore possesses two distinct enzymes: a sensitive chromosome-encoded enzyme and a resistant plasmid-encoded enzyme.
Rarely, sulphonamides cause Stevens–Johnson syndrome
Sulphonamides are relatively free of toxic side effects, but rashes and bone marrow suppression can occur.
Trimethoprim (and co-trimoxazole)
Trimethoprim is a structural analogue of the aminohydroxypyrimidine moiety of folic acid and prevents the synthesis of THFA
Trimethoprim is one of a group of pyrimidine-like molecules analogous in structure to the aminohydroxypyrimidine moiety of the folic acid molecule (Fig. 33.21). Other agents with a similar structure and mechanism of action include the antimalarial pyrimethamine and the anticancer drug methotrexate.
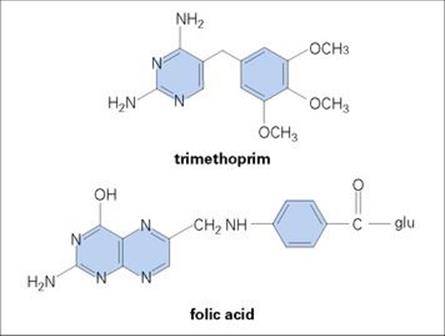
Figure 33.21 Trimethoprim resembles the aminohydroxypyrimidine moiety of folic acid and in this way antagonizes the enzyme dihydrofolate reductase.
Trimethoprim, like sulphonamides, prevents THFA synthesis, but at a later stage by inhibiting dihydrofolate reductase (Fig. 33.20). This enzyme is present in mammalian cells as well as bacterial and protozoan cells, and selective toxicity depends upon the far greater affinity of trimethoprim for the bacterial enzyme.
Trimethoprim is often given in combination with sulphamethoxazole as co-trimoxazole. The advantages of this combination over either drug alone are:
• Mutant bacteria resistant to one agent are unlikely to be resistant to the other (i.e. double mutation).
• The two agents act synergistically against some bacteria (i.e. the combined action of the two bacteriostatic agents has a bactericidal effect that is greater than the action of either agent alone).
Trimethoprim can be given orally (either alone or as co-trimoxazole) or by intravenous infusion (alone or accompanied by sulphonamide). Trimethoprim is excreted in urine, and in patients with severe renal failure it is excreted more rapidly than sulphonamide so that the synergistic ratio of the combination may be lost.
Trimethoprim is often given with sulphamethoxazole as co-trimoxazole for urinary tract infections
Trimethoprim alone is active against Gram-negative rods with the exception of Pseudomonas spp. and its main use is in the treatment (and long-term prophylaxis) of urinary tract infection (see Ch. 20); however, the development of resistance is a concern.
Co-trimoxazole is active against a wide range of urinary tract pathogens and against S. typhi. This combination is also valuable for the treatment of pneumonia caused by the fungus Pneumocystis jirovecii (formerly P. carinii), although pentamidine, another pyrimidine derivative, is probably the preferred alternative (see Ch. 30). Co-trimoxazole is also useful for the treatment of nocardiosis (see Ch. 30) and chancroid (see Ch. 21).
Resistance to trimethoprim is provided by plasmid-encoded dihydrofolate reductases
Plasmid-encoded dihydrofolate reductases with altered affinity for trimethoprim allow the synthesis of THFA to proceed unhindered by the presence of trimethoprim. The ‘replacement enzymes’ are approximately 20 000-fold less susceptible to trimethoprim while retaining their affinity for the normal substrate. Bacteria that are resistant to sulphonamide and trimethoprim are also resistant to co-trimoxazole.
People with AIDS seem to be more prone to the side effects of trimethoprim and co-trimoxazole
Trimethoprim alone and in combination with sulphamethoxazole can cause neutropenia. Nausea and vomiting may occur.
Other agents that affect dna
Nitroimidazoles
Metronidazole is a nitroimidazole with antiparasitic and antibacterial properties
After entry into the microbial cell, the molecule is activated by reduction, and the reduced intermediate products are responsible for antimicrobial activity, probably through interaction with, and breakage of, the cell’s DNA. The reactive intermediates are short-lived and decompose to non-toxic inactive end-products. Metronidazole is active only against anaerobic organisms because only these can produce the low redox potential necessary to reduce the parent drug.
Metronidazole has also been used as a hypoxic cell sensitizer in radiotherapy.
Metronidazole is usually given orally or rectally. It is well absorbed and well distributed in tissues and CSF. The drug is metabolized and most of the parent compound and metabolites are excreted in the urine.
Metronidazole was originally introduced for the treatment of the flagellate parasite Trichomonas vaginalis
Metronidazole is also effective against other protozoan parasites such as Giardia intestinalis and Entamoeba histolytica. It is an important agent for the treatment of infections caused by anaerobic bacteria.
Metronidazole resistance is of increasing concern in T. vaginalis, G. intestinalis, and several anaerobic and microaerophilic bacteria, and commonly involves either an alteration in uptake or a decrease in cellular reductase activity, thereby slowing the activation of the intracellular drug.Helicobacter pylori, a microaerophilic bacterium causing ulcers and gastritis, has been frequently treated with metronidazole. However, resistance can rapidly develop.
Rarely, metronidazole causes central nervous system side effects
The most serious side effects of metronidazole involve the central nervous system and include peripheral neuropathy. However, these are relatively uncommon and usually seen only in patients on large doses or prolonged treatment.
Inhibitors of cytoplasmic membrane function
The cytoplasmic membranes that encompass all kinds of living cells perform a variety of vital functions. The structure of these membranes in bacterial cells differs from that in mammalian cells and allows the application of some selectively toxic molecules, but these are few in number compared with those acting at other target sites.
Lipopeptides
Lipopeptides are a new class of membrane-active antibiotics
Daptomycin is a lipopeptide antibiotic with bactericidal activity against a wide variety of Gram-positive bacteria including vancomycin-resistant E. faecalis and E. faecium and methicillin-resistant Staph. aureus and Staph. epidermidis (Fig. 33.22). The compound acts in a calcium-dependent matter to insert and depolarize the bacterial cytoplasmic membrane. This action leads to a number of consequences including the inability to synthesize ATP and interference with uptake of nutrients. At present, resistance to daptomycin has been rare.
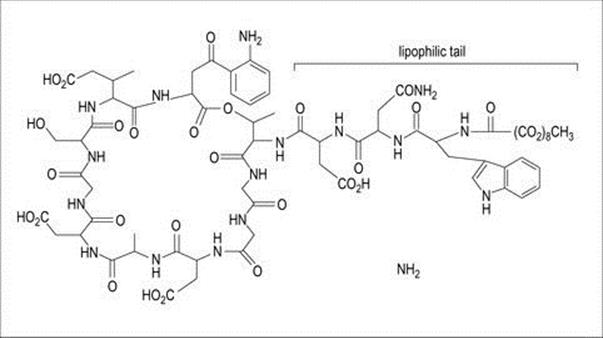
Figure 33.22 Chemical structure of the cyclic lipopeptide, daptomycin, consisting of a 13-member amino acid cyclic lipopeptide with a lipophilic tail which attacks the bacterial cell membrane, causing depolarization and a potassium iron efflux.
Polymyxins
Polymyxins act on the membranes of Gram-negative bacteria
In addition to the polymyxins, the polyene antifungal agents (amphotericin B, nystatin) also act by inhibiting membrane function (see below). Polymyxins are bactericidal cyclic polypeptides that disrupt the structure of cell membranes.
The free amino groups of polymyxins act as cationic detergents, disrupting the phospholipid structure of the cell membrane. Polymyxin B is the most common member of the family still in clinical use.
In the past, polymyxins have been used systemically, but due to poor distribution in tissues, neurotoxicity and nephrotoxicity, they have been superseded by less toxic agents.
Polymyxins are primarily used topically but have also been used in the past for gut decontamination, wound irrigation and as a bladder washout
Polymyxins are active against most Gram-negative organisms except Proteus spp. They are primarily used topically in ointments. After oral administration, polymyxins are not absorbed from the gut, and polymyxin E (colistin) has been used in some gut decontamination regimens for neutropenic patients, although with caution due to concerns regarding renal toxicity. Concerns regarding the lack of effective antibiotics for treating multidrug resistant Gram-negative bacteria have led to renewed interest in polymixin/colistin combination therapy.
Resistance is due to chromosomally mediated alterations in membrane structure or antibiotic uptake.
Urinary tract antiseptics
Nitrofurantoin and methenamine inhibit urinary pathogens
Nitrofurantoin and methenamine are both synthetic compounds that, when taken orally, are absorbed and excreted in the urine in concentrations high enough to inhibit urinary pathogens. Nitrofurantoin has activity only in acid urine. Methenamine is hydrolysed at acid pH to produce ammonia and formaldehyde; it is the formaldehyde that has the antibacterial activity. Nitrofurantoin is used to treat uncomplicated urinary tract infection, and both agents are used to prevent recurrent urinary tract infections. While resistance rarely develops in susceptible bacterial populations, resistance to nitrofurantoin prior to treatment is a concern.
Antituberculosis agents
M. tuberculosis and other mycobacterial infections need prolonged treatment
The treatment of infections caused by M. tuberculosis and other mycobacteria presents an enormous challenge to medicine and the pharmaceutical industry because these organisms:
• have a waxy outer layer that makes them naturally very impermeable and difficult to penetrate with antibiotics
• have an intracellular location, often in cells surrounded by a mass of caseous material, that also makes it difficult for antibiotics to get to them
• grow and multiply extremely slowly, and effective inhibition (and therefore cure) takes weeks or months to achieve. Long-term therapy is therefore a challenge for drug delivery, and orally administrable drugs are consequently highly desirable. It also follows that the emergence of resistance among the mycobacteria and toxicity in the patient are more likely than with the ‘short sharp shock’ treatment more often administered for bacterial infections
• are common and increasing in the wake of the AIDS epidemic in resource-poor countries, where the cost of drug treatment can be prohibitive.
The drugs for first-line therapy of tuberculosis are isoniazid, ethambutol, rifampicin, pyrazinamide and streptomycin
Treatment regimens vary between countries but, with susceptible strains, a 9-month course of isoniazid and rifampicin is an approach that has been used with good success. Pending results of susceptibility tests, a three- or four-drug combination is the common initial treatment, and this is continued for resistant isolates. The structure and mechanism of action of rifampicin and streptomycin have been described in preceding parts of this chapter.
Isoniazid
Isoniazid inhibits mycobacteria and is given with pyridoxine to prevent neurologic side effects
Isoniazid is isonicotinic acid hydrazide, a compound that inhibits mycobacteria, but does not affect other species of bacteria or humans to any great extent. Its bactericidal activity results from inhibition of mycolic acid synthesis, which also accounts for its specificity. It is well absorbed after oral administration, and a single daily dose is usually prescribed except in more difficult cases such as meningitis or miliary tuberculosis. The main toxic effects in humans are neurologic complications, which can be prevented by the concurrent administration of pyridoxine, and hepatitis.
Ethambutol
Ethambutol inhibits mycobacteria, but can cause optic neuritis
Ethambutol is a synthetic molecule that inhibits, but does not kill, mycobacteria. It acts by inhibiting the polymerization of arabinoglycan, a critical constituent of the mycobacteria cell wall. It is well absorbed after oral administration and well distributed in the body, including the CSF. Resistance appears fairly rapidly if the drug is used alone. Thus, it is combined with other drugs in antituberculosis therapy. An important toxic side effect is optic neuritis, and visual acuity should be monitored during therapy.
Pyrazinamide
Pyrazinamide is a synthetic analogue of nicotinamide which appears to target mycolic acid synthesis. After oral administration, the drug is readily absorbed from the gastrointestinal tract and well distributed in body tissues and fluids. It is primarily metabolized in the liver and excreted by the kidney. As with ethambutol, resistance during monotherapy requires that the drug be used in combination with other first-line agents. The most important toxic side effect of pyrazinamide is hepatotoxicity.
Mycobacterial resistance
Drug resistance and immunocompromised patients complicate tuberculosis therapy
Despite the use of antibiotics in combination, the incidence of resistance among mycobacteria is a persistent and increasing problem. Infections with mycobacteria other than M. tuberculosis are on the increase as opportunist infections in people with AIDS, and these organisms tend to be innately more resistant than M. tuberculosis.
Treatment of leprosy
The development of resistance during dapsone monotherapy for leprosy has led to its use in combination with rifampicin
Infection caused by M. leprae is characterized by persistence of the organism in the tissues for years and necessitates very prolonged treatment to prevent relapse. For many years dapsone, related to the sulphonamides (see Fig. 33.19), has been used. This drug has the advantages that it is given orally and it is cheap and effective. However, widespread monotherapy has resulted in the emergence of resistance, and a combination of dapsone, rifampicin and clofazimine, a phenazine compound is now commonly used as multidrug therapy.
Antibacterial agents in practice
It is clear from the preceding sections of this chapter that although there are certain ‘rules of thumb’ about the resistance of bacteria to an antibiotic, it is often impossible to do more than guess in the absence of laboratory tests. Susceptibility tests performed in the laboratory examine the interaction between antibiotics and bacteria in an isolated and rather artificial fashion. At best, the results are a helpful guide to the likely outcome of therapy; at worst, they are misleading. Patient factors such as age, underlying disease, site and type of infection, renal and liver impairment, and drug pharmacodynamics must be taken into account in the antibiotic management of an infection.
Susceptibility tests
Laboratory tests for antibiotic susceptibility fall into two main categories:
• diffusion tests
• dilution tests.
Diffusion tests involve seeding the organism on an agar plate and applying filter paper disks containing antibiotics
The isolate to be tested is seeded over the entire surface of an agar plate, and filter paper disks containing the antibiotics are applied. After overnight incubation the plate is observed for zones of inhibition around each antibiotic disk (Fig. 33.23). The amount of antibiotic in the disk is related to, among other things, the achievable serum concentration and therefore differs for different antibiotics. In addition, antibiotics differ in their ability to diffuse in agar, so the size of the inhibition zone (and not simply its presence) is an indicator of susceptibility of the isolate. The zone sizes are compared with those for reference organisms (either tested in parallel or established previously and published in reference tables) and the result recorded as ‘S’ (susceptible), ‘I’ (intermediate) or ‘R’ (resistant). An ‘I’ result indicates that the isolate is less susceptible than the norm, but may respond to higher doses of antibiotic or in sites where the antibiotic is concentrated (e.g. in urine in the bladder for antibiotics excreted by the kidneys).
Figure 33.23 The antibiotic susceptibility of an organism can be tested by the application of filter paper impregnated with antibiotic onto a lawn of the organisms seeded on an agar plate. After overnight incubation the organism grows and the antibiotics diffuse to produce a zone of inhibition that indicates the degree of susceptibility: disk susceptibility test indicating sulphonamide resistance. SF100 is the sulphonamide disk. (Courtesy of D.K. Banerjee.)
A dilution test provides a quantitative estimate of susceptibility to an antibiotic
A more quantitative estimate of the susceptibility of an organism to an antibiotic can be achieved by performing a MIC (minimum inhibitory concentration) test (i.e. a test to find the lowest concentration that will inhibit visible growth of the bacterial isolate in vitro). Serial dilutions of the test antibiotic are prepared in broth or agar medium and inoculated with a suspension of the test organism. After overnight incubation, the MIC is recorded as the highest dilution in which there is no macroscopic growth (Fig. 33.24). These tests can be performed in a microtitre plate format and form the basis of some automated susceptibility test systems. An alternative approach is the E-test in which a filter paper strip impregnated with a gradient of antibiotic is laid on an agar plate seeded with the test isolate. The concentration on the strip at which growth is inhibited indicates the MIC.
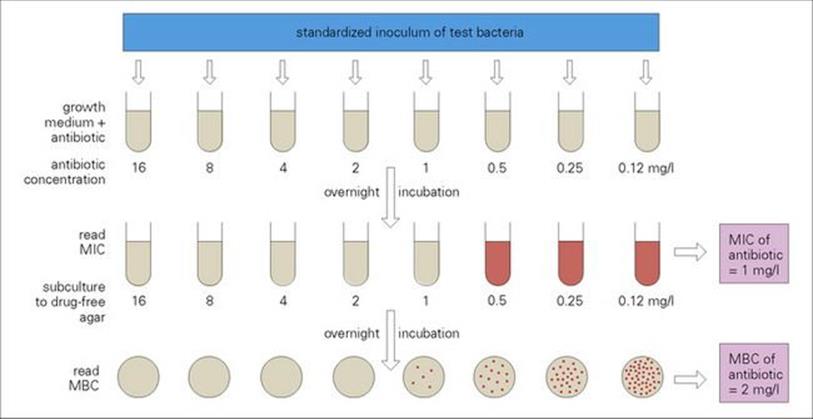
Figure 33.24 More precise measures of the amount of antibiotic required to inhibit and kill a bacterial population can be estimated by establishing the minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) of the antibiotic. Using the standard method as outlined in this illustration, the MIC result is available after 24 h and the MBC result after 48 h. A number of variables such as the inoculum size, the growth medium and the interpretation of the results affect the outcome of MIC tests.
MIC tests are clearly more costly than diffusion tests in terms of time and materials and are not required or used for every isolate from every patient, but they yield useful information for the management of difficult infections or for patients who are failing to respond to apparently appropriate therapy.
An advantage of an MIC test is that it can be extended to determine the MBC (minimum bacterial concentration), which is the lowest concentration of an antibiotic required to kill the organism. In order to discover whether the agent has actually killed the bacteria rather than simply inhibited their growth, the test dilutions are subcultured onto a fresh drug-free medium and incubated for a further 18–24 h (Fig. 33.24). The antibacterial agent is considered to be bactericidal if the MBC is equal to or not greater than fourfold higher than the MIC.
Killing curves provide a dynamic estimate of bacterial susceptibility
One of the disadvantages of MIC and MBC tests is that the result is read at only one point in time. A more dynamic estimate of bacterial susceptibility can be gained by measuring the decrease in viability of the population with time (Fig. 33.25). As with MIC tests, it is not feasible to perform killing curves manually for every test isolate, but they can provide useful information for difficult treatment problems. A number of the automated susceptibility test systems use a measure of bacterial viability (e.g. turbidity, electrical impedance) in the presence of an antibacterial as their indicator system. These machines can produce results more rapidly (e.g. within a few hours) than conventional susceptibility tests. However, automated systems do not work well with fastidious organisms (e.g. S. pneumoniae, N. meningitidis, etc.) or with resistance that is characteristically difficult to detect (e.g. borderline oxacillin MICs in Staphylococcus aureus, ESBLs in Gram-negative isolates, etc.).
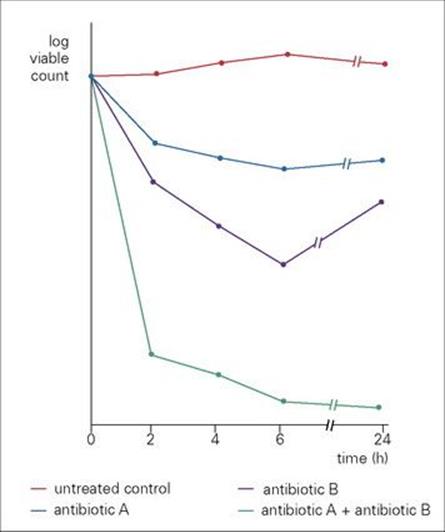
Figure 33.25 A more dynamic picture of the interaction between an antibiotic and a bacterial population can be gained from producing killing curves. In these experiments a culture of 2 × 106 colony forming U/mL was treated with antibiotics A and B alone and in combination. Compared with the untreated control, both A and B inhibit the growth of the bacterial culture, but B is more active than A. However, in combination, the activity of A plus B is synergistic (i.e. it is more active than the sum of the activities of the two antibiotics alone). The combination also prevents the re-growth seen after 6–24 h when the antibiotics are used singly.
Combining antibacterial agents can lead to synergism or antagonism
Hospital patients frequently receive more than one antibacterial agent, and these agents may interact with each other (and also with other drugs such as diuretics).
Antibacterial combinations are described as:
• ‘synergistic’ if their activity is greater than the sum of the individual activities
• ‘antagonistic’ if the activity of one drug is compromised in the presence of the other.
Both diffusion and dilution tests allow the action of combinations of antibiotics to be studied. Although synergy can often be demonstrated in vitro (Fig. 33.26), it is difficult to confirm in vivo. Co-trimoxazole is an example of a combination that is frequently used (see above). Another example is the combination of penicillin (or ampicillin) with gentamicin in the treatment of endocarditis caused by Enterococcus spp., as this combination has been shown to be clearly superior to the effect of the beta-lactam alone (Box 33.6).
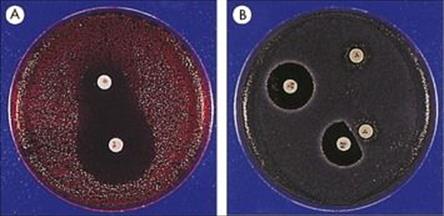
Figure 33.26 (A) Synergy of two antibacterials. Disks containing sulphonamide and trimethoprim have been placed to demonstrate the synergistic activity of these two agents against E. coli. Synergy can be recognized by the fact that the zones of inhibition become continuous between the two disks. (B) Antagonism. Nitrofurantoin is capable of antagonizing the activity of nalidixic acid. When the disks are placed far apart, nalidixic acid inhibits the test organism, but when placed close together this inhibition is antagonized by the presence of nitrofurantoin, as demonstrated by the foreshortening of the zone of inhibition.
![]()
Box 33.6 Use of Antibiotic Combinations
Reasons for using antibiotic combinations. Ideally, single drugs are used, but antibiotic combinations are justifiable under certain circumstances:
• to obtain a synergistic effect, e.g. co-trimoxazole
• to prevent or delay emergence of persistent organisms, e.g. isoniazid, rifampicin and ethambutol for tuberculosis
• to treat polymicrobial infections, e.g. intra-abdominal abscesses where the different microbes have different susceptibilities
• to treat serious infection in the stage before the infectious agent is identified.
![]()
Antagonism can be demonstrated between some pairs of antibiotics in vitro but is rarely evident in vivo.
Antibiotic assays
In the preceding parts of this chapter, the pharmacokinetic properties (absorption, distribution, excretion) of antibacterial agents have been summarized. Some antibacterials have a narrow ‘therapeutic index’, i.e. the concentration required for successful treatment and the concentration toxic to the patient are not very different. The concentrations of such antibiotics should be monitored both to prevent toxicity and to ensure that therapeutic concentrations are achieved. Other less toxic agents should be monitored in some circumstances in some patients (Box 33.7). Serum concentrations are usually measured, but urine, CSF and other body fluids can be assayed if applicable.
![]()
Box 33.7 Importance of Antibiotic Assays
Assays of antibiotics in clinical practice are particularly important when the antibiotic is potentially toxic, but there is a variety of other situations in which assays are important:
• when an antibiotic has a narrow therapeutic index, e.g. aminoglycosides
• when the normal route of excretion of antibiotic is impaired, e.g. in patients with renal failure for agents excreted via the kidney
• when the absorption of the antibiotic is uncertain, e.g. after oral administration
• to ascertain concentrations in sites of infection into which penetration of antibiotics is irregular or unknown, e.g. in cerebrospinal fluid
• in patients receiving prolonged therapy for serious infections, e.g. endocarditis
• in neonates with serious infections
• in patients who fail to respond to apparently appropriate therapy
• to check on patient compliance.
![]()
Antibiotic assays may be performed by a variety of methods such as high-performance liquid chromatography and direct assays for biological activity (bioassay). However, the most common approach uses immunologic methods which can be automated. In this method, the antibiotic in the patient specimen is an ‘antigen’ that competes with a specific level of labelled ‘tracking’ antibiotic for binding sites on an ‘anti-drug’ antibody. Thus, increased antibiotic levels in a patient sample result in decreased binding of tracking antibiotic, etc. Such assays are rapid, require only small volumes of serum, and are highly specific. However, they are obviously only applicable to instances where specific anti-drug antibody is available.
Antiviral therapy
The last two decades have seen a range of new antiviral agents licensed for use against a number of virus infections, including HIV, hepatitis B (HBV), hepatitis C (HCV), herpesviruses and influenza A and B (Fig. 33.27). The current antivirals for treating individuals with virus infections are all virustatic rather than virucidal, in other words, they do not kill viruses but suppress their replication. Of the increasing array of antiviral agents licensed for treatment, more than half are used in the management of HIV-infected individuals. As of 2011, there were 23 antiretroviral agents, excluding the fixed-dose combination agents, that constitute six different classes of drug (summarized in Table 33.7)
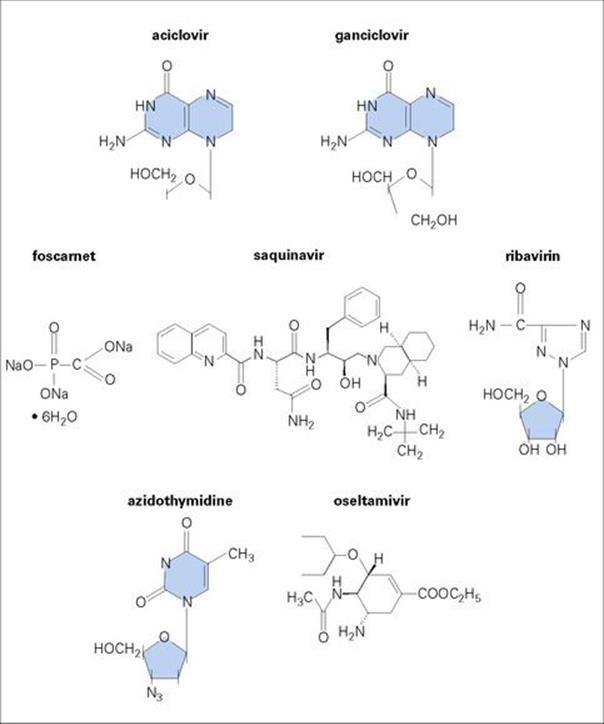
Figure 33.27 Antiviral agents are few in number and narrow in their spectrum of activity, e.g. amantadine is effective against influenza A, but not influenza B, while acyclovir is effective against herpes simplex virus (HSV) and varicella-zoster virus (VZV), but not cytomegalovirus (CMV) or Epstein–Barr virus (EBV).
Table 33.7 Antiviral drugs
|
HIV |
|
|
Nucleoside reverse transcriptase inhibitor (NRTI) |
Zidovudine (AZT) |
|
Didanosine (ddI) |
|
|
Stavudine (d4T) |
|
|
Lamivudine (3TC) |
|
|
Abacavir (ABC) |
|
|
Emtricitabine (FTC) |
|
|
Abacavir/Lamivudine/Zidovudine (Trizivir) |
|
|
Lamivudine/Zidovudine (Combivir) |
|
|
Nucleotide Reverse Transcriptase Inhibitor (NRTI) |
Tenofovir (TDF) |
|
Combinations |
|
|
NRTI |
Tenofovir/Emtricitabine (Truvada) |
|
NRTI and NNRTI |
Tenofovir/Emtricitabine/Efavirenz (Atripla) |
|
Non Nucleoside Reverse Transcriptase Inhibitor (NNRTI) |
Nevirapine |
|
Efavirenz |
|
|
Delavirdine |
|
|
Etravirine |
|
|
Protease inhibitors |
Indinavir |
|
Nelfinavir |
|
|
Ritonavir |
|
|
Saquinavir |
|
|
Amprenavir |
|
|
Lopinavir/Ritonavir (Kaletra) |
|
|
Atazanavir |
|
|
Fosamprenavir |
|
|
Tipranavir |
|
|
Darunavir |
|
|
Fusion inhibitor |
Enfurvitide |
|
Integrase inhibitor |
Raltegravir |
|
Chemokine co-receptor inhibitor |
Maraviroc |
|
HCV |
|
|
Ribavirin |
|
|
Boceprivir |
|
|
Telaprevir |
|
|
Pegylated interferon alpha 2a |
|
|
Pegylated interferon alpha 2b |
|
|
Combination |
Ribavirin/Pegylated interferon alpha 2b |
|
HBV |
|
|
Lamivudine |
|
|
Adefovir |
|
|
Entecavir |
|
|
Telbivudine |
|
|
Emtricitabine |
|
|
Tenofovir |
|
|
Pegylated interferon alpha 2a |
|
|
Herpesviruses: HSV, VZV |
|
|
Acyclovir |
|
|
Valaciclovir |
|
|
Famciclovir |
|
|
Penciclovir |
|
|
Idoxuridine |
|
|
Trifluridine |
|
|
Brivudin |
|
|
CMV including HSV and VZV |
|
|
Ganciclovir |
|
|
Valganciclovir |
|
|
Foscarnet |
|
|
Cidofovir |
|
|
Influenza A and B viruses |
|
|
Neuraminidase inhibtor |
Oseltamivir |
|
Neuraminidase inhibtor |
Zanamivir |
|
Influenza A viruses |
|
|
Amantadine |
|
|
Rimantadine |
|
|
RSV (also can be used in measles, Lassa fever, adenovirus and HCV infections) |
|
|
Ribavirin |
|
The problem in developing new antivirals has been mostly due to the difficulty of interfering with viral activity in the cell without adversely affecting the host. This is because viruses are dependent on the host cell’s protein synthetic machinery. Directions taken in the development of new antivirals include:
• Adsorption inhibitors
• CCR5 and CXCR4 antagonists that target HIV-1 attachment, co-receptor binding and fusion referred to as entry inhibitors
• Blocking viral mRNA with short nucleotide sequences that are complementary to viral sequences. These antisense oligonucleotides bind to newly transcribed viral RNA and block its action
• Using compounds that inactivate virus when used topically.
Reports have highlighted the importance of making an early diagnosis in short-incubation-period viral infections, such as influenza, in order for antiviral treatment to be successful. Moreover, virus-specific replication steps can be identified (Fig. 33.28), and more of these will doubtless be exploited, such as identifying virus-induced enzymes. In addition, apart from improving the ease of administration of those drugs where the pill burden is high, as in HAART, the lack of therapeutic options for a number of viral infections, including human papillomaviruses and adenovirus infections, is an area for further research and development.
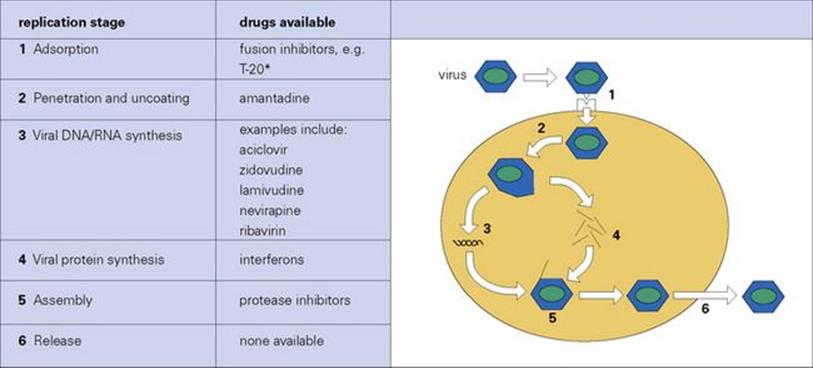
Figure 33.28 The site of action of antiviral agents. Resistance to agents is uncommon, but does occur (e.g. cytomegalovirus strains resistant to ganciclovir, herpes simplex virus strains resistant to acyclovir). Adsorption of virus to cell can be blocked by virus-specific antibody. * T-20 binds to the HIV gp41 site, preventing attachment to the T cell.
Bearing in mind that antivirals can be used to treat acute and chronic viral infections, and in the latter case may be given for many years or for life, considerations include the length of the treatment course, single versus combination therapy, drug pharmacokinetics and interactions, adverse effects and antiviral resistance. Monitoring viral load as a marker of prognosis and treatment response is important in chronic viral infections such as HIV, HBV and HCV, together with therapeutic drug monitoring and genotypic and phenotypic resistance tests.
Antiviral resistance occurs with varying prevalence in different patient populations: for example, acyclovir-resistant HSV and ganciclovir-resistant CMV are mostly seen in immunocompromised individuals at a low level. Antiretroviral resistance is seen across all the main classes of agents – nucleoside reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors, and protease inhibitors – with increasing frequency in resource-rich countries. Lamivudine-resistant HBV is well recognized and is usually detected after a couple of years of treatment. Drug resistance involving most of the other agents used to treat HBV carriers also occurs. One issue with antiviral resistance is that the replication fitness of the drug-resistant variants is often less than the wild-type strain. In addition, in the case of a number of viruses, including HBV and HCV, the response varies depending on the viral genotype.
Some viral infections have an immunopathologic basis, such as CMV pneumonitis, in which case an antiviral is given in combination with an immunoglobulin preparation. This may be human normal immunoglobulin or virus-specific immunoglobulin, i.e. CMV hyperimmune globulin. Moreover, an immunomodulator may be given in conjunction with an antiviral such as pegylated interferon and ribavirin to treat hepatitis C virus infection.
Palivizumab is an example of a humanized monoclonal antibody produced to prevent infection. It is directed against the respiratory syncytial virus (RSV) fusion protein and has potent neutralizing and fusion inhibitory activity. It is used in specific clinical settings to prevent severe lower respiratory tract infections caused by RSV requiring hospitalization in children born at 35 weeks’ gestation or less who are less than 6 months old at the onset of the RSV season. In addition, it may be used in children less than 2 years of age with specific respiratory and cardiac conditions such as bronchopulmonary dysplasia.
Finally, in the case of some viral respiratory tract infections, antibiotics are often given to control or act as prophylaxis against a secondary bacterial infection. Influenza infection is an example where staphylococcal and streptococcal pneumonia may occur after the initial virological insult.
It is difficult to group the antiviral drugs in the same way as the antibiotics. One can either look at them as, for example, anti-HIV, anti-HBV and anti-HCV or group them under mechanism of action. The following are classified using the latter heading.
Prodrugs that target the viral DNA polymerase
Acyclovir, valaciclovir, penciclovir, famciclovir, ganciclovir, valganciclovir, cidofovir.
Acyclovir (acycloguanosine)
Acyclovir inhibits HSV and varicella-zoster virus (VZV) DNA polymerase
Acyclovir is used in the treatment of HSV and VZV infections. A number of other agents include valaciclovir, the l-valyl ester of acyclovir, and famciclovir. Acyclovir is inactive until phosphorylated and is an example of a prodrug. Acyclovir (Fig. 33.29) is phosphorylated by the herpesvirus thymidine kinase and the monophosphate is then converted by cellular kinases to the triphosphate, which inhibits the herpesvirus DNA polymerase. As it is taken up and efficiently phosphorylated by HSV-infected cells, the action on cellular DNA polymerase is minimal and toxic side effects such as neutropenia and thrombocytopenia are rare. The drug is also incorporated into viral DNA, resulting in chain termination. As it is excreted by the kidney, the drug can crystallize in the renal tract in individuals with renal failure, causing acute tubular necrosis. Otherwise, acyclovir has an excellent safety profile.
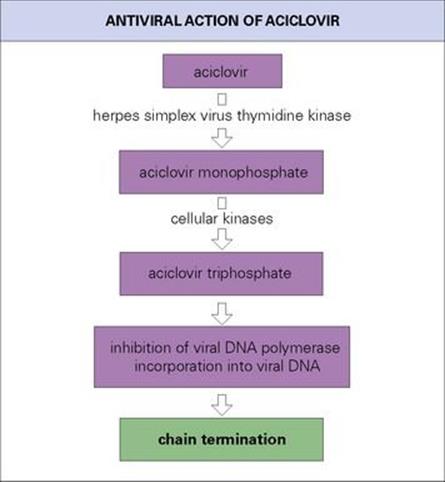
Figure 33.29 The activity of an antiviral agent against different herpes viruses is correlated with the ability of the viruses to induce a thymidine kinase; hence acyclovir is most active against herpes simplex virus and least active against cytomegalovirus.
Systemic acyclovir revolutionized the treatment of HSV encephalitis, and HSV and VZV infections in immunocompromised patients. It is effective in treating primary and recurrent genital herpes. In shingles (herpes zoster), recovery is accelerated and post-zoster pain reduced. As with HSV, the varicella-zoster virus remains latent in ganglia and can reactivate.
As the oral bioavailability is only 15–20%, acyclovir is given intravenously in a number of clinical settings initially. Valaciclovir and famciclovir have improved bioavailability profiles in comparison with acyclovir, resulting in less frequent daily dosages.
Ganciclovir (dihydroxypropoxy-methylguanine, DHPG)
Ganciclovir is structurally similar to acyclovir but has an extra hydroxyl group. The range of activity is broader than that of acyclovir, and the drug is active against CMV infections. CMV does not encode a thymidine kinase, but the drug is monophosphorylated by a virus UL97 gene-specified kinase and then further phosphorylated by cellular kinases. However, selective toxicity is not seen, and it is myelosuppressive, its main adverse effect being bone marrow toxicity. Ganciclovir triphosphate inhibits CMV DNA polymerase. It is given intravenously because of limited oral bioavailability. However, an oral agent, valganciclovir, has improved the outpatient management of individuals with CMV infections, as it has equivalent activity to intravenous ganciclovir.
Ganciclovir is given to treat CMV retinitis, encephalitis and gastrointestinal disease seen in immunocompromised individuals. It is also used as pre-emptive therapy in bone marrow transplant as well as solid organ transplant recipients, who are monitored regularly for the presence of CMV in their blood as this leads to CMV dissemination.
Valganciclovir
Valganciclovir is the valine ester of ganciclovir, has similar bioavailability but has the advantage of being given orally.
Cidofovir (HPMPC)
Cidofovir is another chain terminator that targets the viral DNA polymerase. It is phosphorylated intracellularly to the diphosphate form and is then added to the 3′ end of the viral DNA chain. It is effective against CMV and has been used to treat adenovirus infections. When given topically or intralesionally, it has activity against genital warts and can be used to treat acyclovir-resistant HSV infections. It has to be given intravenously and is nephrotoxic.
Pyrophosphate analogue that blocks the pyrophosphate binding site on the viral DNA polymerase
Foscarnet (phosphonoformate)
This compound attaches to the pyrophosphate-binding site of the herpesvirus DNA polymerase, preventing nucleotide binding and therefore inhibiting viral replication. It is used in treating CMV infections and is active against HSV and VZV and can be used to treat acyclovir-resistant HSV infections. It is nephrotoxic and is often used as a second-line agent.
Nucleoside and nucleotide reverse transcriptase inhibitors (NRTIs)
The aim of antiretroviral therapy is to lower and keep the plasma HIV-1 RNA load below the limit of assay detection and therefore maintain the CD4 count. There are a number of treatment guidelines, examples of which can be found in the bibliography: zidovudine (azidothymidine, AZT), didanosine (ddI, deoxyinosine), lamivudine (3TC, thiacytidine), stavudine (d4T, didehydrodideoxyuridine), abacavir, emtricitabine, and tenofovir. These drugs have similar modes of action and are mostly used in conjunction with the other main classes of antiretroviral drugs: the non-nucleoside reverse transcriptase inhibitors and protease inhibitors to treat HIV-1-infected individuals.
Zidovudine (azidothymidine, AZT)
Zidovudine is an analogue of the nucleoside thymidine in which the hydroxyl group on the ribose is replaced by an azido group. After conversion to the triphosphate by cellular enzymes (Fig. 33.30) it acts as an inhibitor of, and substrate for, the viral reverse transcriptase. The azido group prevents the formation of phosphodiester linkages. Proviral DNA formation is blocked because AZT triphosphate is incorporated into the DNA, with resulting chain termination.
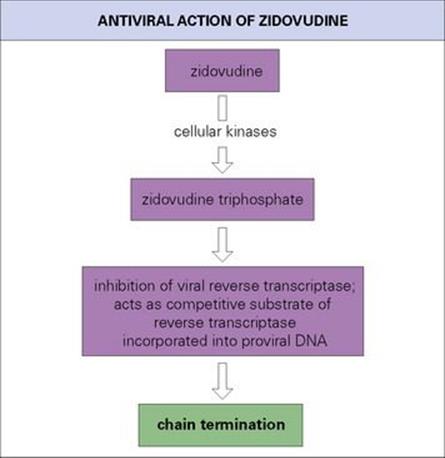
Figure 33.30 HIV reverse transcriptase is 100 times more sensitive than host cell DNA polymerase to zidovudine triphosphate, but toxic effects are not uncommon.
Zidovudine is given orally. Toxicity is a problem, with bone marrow suppression (macrocytic anaemia, neutropenia, leucopenia) and less commonly nausea, vomiting, headache, myalgia and malaise. This was more often seen in the early days of HIV treatment when the drug was given at a high dose. Other adverse events include lactic acidosis, hyperlipidaemia, lipoatrophy and insulin resistance or diabetes mellitus. Regular blood tests are necessary to detect anaemia and myelosuppression.
Like zidovudine, the other nucleoside analogues are converted to triphosphates and inhibit the HIV reverse transcriptase. Some of these agents have been combined as fixed-dose treatments such as combivir (AZT and 3TC), truvada (emtricitabine and tenofovir), and trizivir (AZT, 3TC and abacavir).
There are a number of adverse effects shared by this class of drugs but the more specific side effects include pancreatitis (ddI), peripheral neuropathy (d4T, ddI), lipodystrophy, i.e. fatty tissue redistribution from subcutaneous areas such as the face and limbs, to the neck and abdominal viscera (d4T), and hypersensitivity (abacavir). Mitochondrial toxicity due to inhibition of the mitochondrial DNA polymerase and lactic acidosis is also reported.
Drug resistance can lead to cross-resistance to other nucleoside analogues.
Tenofovir is a nucleotide reverse transcriptase inhibitor and is phosphorylated to the diphosphate form that acts as chain terminator.
The nucleoside and nucleotide RTIs and most of the protease inhibitors can be used to treat HIV-2-infected individuals. The non-nucleoside RTIs cannot be used and the fusion inhibitor, enfuvirtide, has reduced HIV-2 activity.
Non-nucleoside reverse transcriptase inhibitors (NNRTIs)
Nevirapine, efavirenz (EFV), delavirdine and etravirine
These are used in combination with the nucleoside analogues and may be used as first-line drugs before moving to the protease inhibitor class. This is because they lead to a rapid fall in the plasma HIV-1 RNA load, especially in those individuals with very high HIV loads for whom protease inhibitor treatment is being considered, and have fewer side effects. They act as non-competitive inhibitors of HIV-1 reverse transcriptase by binding to a hydrophobic pocket proximal to the enzyme catalytic site. They are inactive against HIV-2. The NNRTIs are inducers of cytochrome P450 and it is important to consider potential drug interactions. The most common adverse effect with nevirapine is a skin rash. Efavirenz may cause vivid dreams and sleep disturbance initially and should not be used in the first trimester of pregnancy.
A single mutation in the reverse transcriptase leads to resistance to these drugs, effectively removing this class of drug from the treatment regimen. In 2006–07, primary NNRTI resistance rates were about 8% in the USA and 2% in Europe.
Protease inhibitors
Nelfinavir, saquinavir, indinavir, ritonavir, lopinavir plus ritonavir (Kaletra), atazanavir, amprenavir, darunavir, fosmaprenavir, tipranavir
The protease enzyme acts in the post-translational cleavage of the gag and gag-pol polyproteins into the structural proteins and enzymes critical for viral replication. The result of protease inhibition is the production of immature, defective viral particles. Protease inhibitors (PIs) were introduced to HIV treatment combinations in 1996 and had a great effect on the control of HIV infection. Their use led to the term highly active antiretroviral therapy (HAART). They are peptidomimetic inhibitors of the viral protease and prevent the cleavage of the gag and gag-polpolyproteins into functional structural proteins and enzymes. They are very potent drugs which lead to a rapid fall in the plasma HIV RNA load, especially in those individuals with very high HIV loads. They are usually given in combination with nucleoside analogues. They are metabolized and excreted rapidly and have to be taken several times daily. Side effects include gastrointestinal disturbances, the lipodystrophy syndrome (body fat redistribution), increased triglycerides, and insulin resistance leading to diabetes.
Drug resistance is well recognized and a number of protease mutations result in cross-resistance. Boosting atazanavir and darunavir with low-dose ritonavir leads to greater virological activity due to improved pharmacodynamics. However, higher rates of side effects are seen. Darunavir and ritonavir is useful for treating patients with PI-resistant HIV. Kaletra is a combination of lopinavir and ritonavir and is associated with sustained viral suppression as well as less drug resistance.
Fusion inhibitors
Enfuvirtide, also known as T-20, is a peptide that blocks HIV before it enters the host cell by competitively binding to gp41, the transmembrane glycoprotein and blocking the post-fusion structure from forming. It therefore should not cross-react with the other classes of antiretroviral drugs. It is given twice daily as a subcutaneous injection and is approved for salvage therapy in those treatment-experienced individuals with resistance mutations to the other drug classes. Adverse events include pain at the injection site and rare hypersensitivity reactions.
Integrase inhibitors
Raltegravir is an HIV integrase strand transfer inhibitor (INSTI). Integration involves transferring virally encoded DNA into the host chromosome. It is a three-step process including the formation of a preintegration viral DNA complex; 3′ processing; and strand transfer. Raltegravir inhibits the strand transfer step. It is thought that it interacts with divalent cations of the catalytic core of the integrase. INSTIs are also active against HIV strains resistant to other classes of antiretroviral agents. Three major resistance mutations have been reported leading to integrase inhibitor resistance. Side effects are mostly gastrointestinal.
Chemokine receptor antagonists
HIV-1 entry into host cells involves the viral envelope protein binding to the CD4 receptor and subsequently to a chemokine co-receptor. Two co-receptors identified are called CCR5 and CXCR4. Tests that identify the viral phenotype have been used to determine the populations of virus in someone with HIV and these are referred to as R5-tropic, X4-tropic or dual/mixed. Diagnostic laboratories use genotypic tests to predict viral co-receptor tropism, R5 or X4, based on the sequence of the viral envelope on the basis of algorithms.
Maraviroc is a CCR5 chemokine co-receptor antagonist and was approved originally for adults who had been given HAART and had R5 HIV-1 infection.
Inosine monophosphate dehydrogenase inhibitor
Ribavirin (tribavirin)
This guanosine analogue is triphosphorylated by cellular enzymes. It has various actions including inhibition of production of guanosine triphosphate pools needed for viral nucleic acid synthesis. Ribavirin can target both RNA and DNA viruses. Once triphosphorylated, it can also interfere with the viral RNA polymerase. It is used clinically as an aerosol for treating severe respiratory syncytial virus (RSV) infection in infants and for arenavirus infections such as Lassa fever (see Ch. 26). Oral ribavirin could be used as postexposure prophylaxis for Lassa fever in the case of high-risk exposure incidents. It is also active against measles virus and hepatitis C virus infection (see below).
Antivirals targeting influenza viruses
Amantadine, rimantadine, zanamivir, oseltamivir, and peramivir. These drugs have selective activity against influenza viruses and so have been grouped together with this header rather than by their mode of action. Amantadine and rimantadine only have activity against influenza A. The neuraminidase inhibitors, zanamivir, oseltamivir, and peramivir have increased the range of activity by inhibiting both influenza A and B viruses.
Amantadine and rimantadine
These drugs specifically inhibit the replication of influenza A viruses, but have no effect on influenza B and other respiratory viruses. They act by inhibiting the penetration of virus into the cell, or its uncoating. Fusion of the viral envelope with a cell membrane, which normally occurs at a low pH, is prevented. Amantadine acts on the viral matrix protein ion channel, thus stopping hydrogen ion passage, raising the pH in intracellular vacuoles, and therefore blocking infection. The standard dose can cause minor neurologic side effects such as insomnia, dizziness and headache, especially in elderly patients, and this has discouraged its widespread use. Amantadine can be given prophylactically during community outbreaks of influenza A. It can also be used for treatment, and if taken within 48 h of symptoms there is a reduction in disease severity. However, rapid emergence of drug-resistant variants can occur, and due to the inactivity against influenza B and central nervous system side effects, and the development of the neuraminidase inhibitors, this class of drugs is of less importance in the influenza armamentarium.
Neuraminidase inhibitors: zanamivir, oseltamivir, and peramivir
Neuraminidase is one of the two surface glycoproteins studded on the influenza virus surface. It cleaves N-acetylneuraminic acid, also known as sialic acid, residues from the host cell, thus releasing the virus and allowing further spread in the respiratory tract.
The neuraminidase inhibitors (NAIs) are N-acetylneuraminic acid analogues and act as competitive reversible inhibitors of the neuraminidase enzyme active site. Zanamivir is an inhaled agent and can be given intravenously, oseltamivir is an oral drug and peramivir is an intravenous agent, all of which are cleaved by esterases to the active carboxylate form and act on influenza A and B. The importance of having an increased armamentarium of NAIs was demonstrated during 2007–08 and 2008–09 as oseltamivir resistance emerged globally amongst the influenza A H1N1 viruses. In the USA, oseltamivir resistance was seen in around 20% and 90% of influenza A H1N1 viruses tested during both the above seasons, respectively.
These drugs reduce viral shedding, disease severity, duration and symptoms if given early in infection and can be used as prophylaxis. They are effective against the circulating influenza strains including the avian influenza H5N1 virus.
Hepatitis B treatment
The aim of treating individuals with chronic hepatitis B and C virus infections is to reduce the risk of cirrhosis and hepatocellular carcinoma by suppressing HBV DNA and HCV RNA levels, respectively.
Treatment regimens offered to hepatitis B virus carriers include nucleoside and nucleotide analogues such as lamivudine, adefovir entecavir, telbivudine and emtricitabine. After stopping treatment, this response may be reversed and continuing treatment long term may lead to the development of antiviral resistance, although the virus will be less fit than the wild type. Lamivudine, emtricitabine and adefovir monotherapy is not recommended.
Immunomodulation using pegylated interferon alpha, which has a longer half life than interferon preparations that do not include polyethylene glycol, enhances the innate immune response by binding to the type 1 interferon receptor. This leads to up-regulation of multiple interferon-stimulated genes limiting viral replication. In hepatitis B e antigen positive and negative carriers, 48 weeks of pegylated interferon alpha results in HBV DNA loss in 25% and 63% of patients, respectively. However, interferons have a large side effect profile.
Of the nucleoside analogues, lamivudine therapy results in improved liver histology and liver enzyme levels in 56% and 72% of patients, respectively. The genetic barrier to resistance is low as only one mutation is needed to lead to lamivudine resistance, compared with entecavir which has a higher barrier as three mutations are needed.
Emtricitabine cannot be used as single-agent therapy due to high rates of resistance. Telbivudine is effective but has a low genetic barrier to resistance.
Adefovir and tenofovir are acyclic diphosphonates with tenofovir being more effective than adefovir. They are prodrugs as they need to be phosphorylated to become active and are analogues of adenosine monophosphate. They affect the HBV polymerase by competitively inhibiting deoxyadenosine 5′-triphosphate, resulting in chain termination. The major side effect is nephrotoxicity.
Entecavir is a guanosine analogue that is one of the more effective drugs. It inhibits the HBV DNA polymerase by preventing the following functions: priming of the HBV DNA polymerase, reverse transcription of the negative strand from the pregenomic messenger RNA, and synthesis of positive-strand HBV DNA.
These oral antiviral agents have changed the treatment landscape in chronic hepatitis B carriers, and the role of combination therapy needs to be investigated.
Hepatitis C treatment
Pegylated interferon alpha combined with ribavirin is the standard treatment of chronic HCV infection. Antiviral treatment may lead to a sustained virological response (SVR) and long-term clinical benefit shown by the serum HCV RNA being below the limit of assay detection 24 weeks after starting treatment. HCV genotype 1 is the most common genotype globally and 48 weeks of the above combination treatment can lead to SVRs of only up to 50%. However, SVRs are seen in 75–90% of infected individuals with HCV genotype 2 and 3 infections.
Determining the liver histology by carrying out a liver biopsy may not be necessary. This is not the case in genotype 1, 4, 5 and 6 infection, although the development of non-invasive markers of liver fibrosis, serological markers and ultrasound techniques reduce the need for a liver biopsy. Identifying the HCV genotype is critical, as it assists the decision as to whether treatment is indicated and the length of the treatment course.
The HCV nonstructural (NS) NS3/4A serine protease is an enzyme that cleaves the HCV-encoded polyprotein leading to mature viral proteins. NS3/4A protease inhibitors include telaprevir, boceprevir and danoprevir and although they can result in improved SVRs, resistance develops rapidly if given as monotherapy. Boceprevir is a competitive inhibitor of the NS3 protease of HCV genotype 1 but does not have clinically significant activity against other HCV genotypes. Studies in which, for example, telaprevir was given in combination with pegylated interferon and ribavirin, showed good SVR rates. Anaemia is a significant side effect with this class of drugs.
Intereferons – immunomodulatory agents
Interferons (see Ch 9) are natural glycoproteins produced by the innate immune system in response to infections. They have a non-virus-specific antiviral and immunomodulatory actions and trigger a cascade of intracellular reactions that activate IFN-inducible genes. These genes encode proteins thought to inhibit intracellular virus multiplication by inhibiting translation initiation and assisting RNA degradation. IFN-alpha also binds to immune cells resulting in class I MHC antigen expression, activation of effector cells and a cytokine cascade. Production of TH1 cells is stimulated, in contrast to TH2 suppressor cells that are reduced. IFNs are generally given as subcutaneous injections and the side effects are significant and include tiredness, headache, myalgia and psychiatric symptoms.
IFNs have been used to treat individuals with chronic HBV and HCV infections and have an effect on human papillomavirus infections, given by intralesional injection, but are not used routinely.
When given in the past as monotherapy, success was limited due to the poor SVR rates for both HBV and HCV infections.
Other targets
Drugs targeting post-translational processing, virus entry, RNA translation and virus assembly and release are being developed in addition to host cell-targeting compounds. Nucleic acid-based antiviral agents including antisense oligonucleotides and RNA interference-based agents have been synthesized for clinical trials. Other potential therapeutic options include immunotherapy using antibody-based preparations and therapeutic vaccines.
Clinical management of antiviral therapy
Viral load and antiviral resistance tests as well as therapeutic drug monitoring assist in clinical management
Qualitative and/or quantitative nucleic acid tests are critical in the diagnosis, treatment decision, assessment of response to treatment and prognosis for a number of viral infections. This is true for HIV load testing, together with the CD4 count and percentage. With HCV, the main determinant of HCV treatment response and duration is the HCV genotype, having determined the plasma HCV RNA load. However, monitoring the HCV RNA level during therapy is not recommended for patients with genotype 2 or 3 infections, as most become HCV RNA negative early during treatment. For HBV infection, plasma HBV DNA load and antiviral resistance testing are part of the clinical management strategy. Genotypic analysis is also helpful. Another example is CMV DNA monitoring in post-transplant populations to detect early viraemia in order for pre-emptive treatment to be given.
The main causes of treatment failure in HIV infection are either compliance issues or the development of antiviral resistance.
Highly active antiretroviral therapy (HAART) has had an enormous impact on HIV disease progression. However, HAART can fail in up to 50% of individuals on treatment, especially if they have failed on previous treatment regimens. The development of drug-resistant virus will lead to treatment failure as seen by an increase in HIV load and reduction in CD4 count. Specific mutations can be detected in the drug target sites, i.e. reverse transcriptase and protease regions, by nucleic acid sequencing. This is referred to as a genotypic resistance assay. Key mutations known as primary resistance mutations at specific codons have been associated with a reduction in susceptibility to the various clans of antiretroviral drugs. Some mutations are unique to certain drugs, but many confer cross-resistance, resulting in a clan of drugs, such as the NNRTIs, being removed from the treatment regimen. In addition, viral tropism assays are carried out in diagnostic laboratories to identify co-receptor use which is critical when deciding on using chemokine receptor antagonists. HIV-1 entry into lymphocytes and monocytes involves binding of the gp120 envelope glycoprotein to the CD4 receptor, followed by interaction with one of two main co-receptors, CCR5 or CXCR4. This is referred to as viral tropism and whether the virus is X4 or R5 is mainly determined by the amino acid sequence of the V3 region of gp120. Dually tropic strains can use both receptors. In later-stage HIV-1 infection, the CD4 cell count falls and the minority population X4 or R5/X4 strains rises within the viral quasispecies and can finally emerge as a majority population. HIV-1 tropism can be determined using phenotypic and genotypic methods. Genotypic tropism testing can be carried out in laboratories and predictions of co-receptor use are based on the amino acid sequence of the gp120 V3 loop using interpretative algorithms. Antiretroviral drug regimens are based on the results of antiretroviral resistance sequencing assays as well as viral tropism assays.
As HIV drug resistance can be transmitted, and the prevalence of resistant viruses is increasing in individuals with a new HIV diagnosis, baseline genotypic resistance testing is very important in order to tailor HAART appropriately. In addition, this is being used more frequently to optimize the treatment regimen during drug failure episodes. Details of the key mutations can be found on specialist HIV websites together with guidelines for managing HIV infected individuals.
It is important in HIV infection to continue the drugs whilst carrying out resistance tests, as without the ‘driver’ there is a reversion to the wild-type strain as the minor viral populations that contain the mutations are deselected. Phenotypic analysis may also be helpful.
The effectiveness of HAART is dependent on good drug plasma concentrations. Keeping drug concentrations within a therapeutic range is critical and drug interactions and compliance issues may result in high or low drug levels, leading to toxicity or virological failure, respectively. Therapeutic drug monitoring is carried out in specialist laboratories and is helpful in finding and correcting any such problems.
Antifungal agents
Compared with antibacterials, the number of suitable antifungal drugs is very limited. Selective toxicity is much more difficult to achieve in the eukaryotic fungal cells than in the prokaryotic bacteria, and although the available antifungals have greater activity against fungal cells than they do against human cells, the difference is not as marked as it is for most antibacterial agents. Treatment of fungal infections is further hampered by problems of solubility, stability and absorption of the existing drugs, and the search for new agents is a high priority. Drug resistance is also increasing.
Antifungals can be classified on the basis of target site and chemical structure
Like antibacterials, antifungals can be classified on the basis of target site and chemical structure. This immediately reveals a major difference between antibacterial and antifungal agents, with the major antifungals acting on the synthesis or function of the intracellular membranes. The exceptions are flucytosine (5-fluorocytosine) and griseofulvin, which interfere with DNA synthesis, and caspofungin, which inhibits cell wall formation. There are currently no inhibitors of fungal protein synthesis that do not also inhibit the equivalent mammalian pathway.
Azole compounds inhibit cell membrane synthesis
Azole antifungals act by inhibiting lanosterol C14-demethylase, an important enzyme in sterol biosynthesis. Clotrimazole and miconazole are useful as topical preparations. Ketoconazole and fluconazole are used in treatment of a variety of serious fungal infections (Table 33.8), and fluconazole is often used in the treatment of Candida infections. Resistance to the azoles is becoming more widespread and threatens to compromise this group of compounds. Newer azole compounds include posaconazole, which is used in aspergillosis unresponsive to amphotericin B, and voriconazole, which is used against invasive candidiasis resistant to fluconazole.
Table 33.8 The major therapeutic applications of antifungal drugs

Echinocandins interfere with cell wall synthesis
The echinocandins caspofungin, micafungin and anidulafungin inhibit the enzyme β-(1,3)-D-glucan synthase which is required for synthesis of an essential part of the fungal cell wall. This important group of compounds offers new therapeutic options against infections such as invasiveAspergillus infections, candidaemia and invasive candidiasis and Pneumocystis. However, they are not active against Cryptococcus neoformans.
Polyenes inhibit cell membrane function
Amphotericin B and nystatin act by binding to sterols in cell membranes, resulting in leakage of cellular contents and cell death. Their preferential binding to ergosterol over cholesterol is the basis for selective toxicity. Amphotericin remains the drug of choice for the treatment of serious systemic fungal infections despite its serious toxic side effects; lipid formulations have lower toxicity and are increasingly preferred. Nystatin is used only in topical formulations.
Flucytosine and griseofulvin inhibit nucleic acid synthesis
Flucytosine (5-fluorocytosine) is deaminated to 5-fluorouracil, which inhibits DNA synthesis. Selective toxicity is based on the preferential uptake by fungal cells compared with host cells. Flucytosine is active only on yeasts (e.g. Candida spp. and Cryptococcus). Resistance emerges rapidly to flucytosine, which should therefore be used in combination with amphotericin B (whereby it is sometimes possible to reduce the dose of amphotericin B and therefore the toxic side effects).
Griseofulvin appears to inhibit nucleic acid synthesis and to have antimitotic activity, possibly by inhibiting microtubule assembly. It may also have effects on cell wall synthesis by inhibiting chitin synthesis. In the host, griseofulvin binds specifically to newly formed keratin and is active in vivo only against dermatophyte fungi (see Chs 4 and 26).
Other topical antifungal agents include Whitfield’s ointment, tolnaftate, ciclopirox, haloprogin and naftifine
A variety of agents such as Whitfield’s ointment (a mixture of benzoic and salicylic acids), tolnaftate, ciclopirox, haloprogin and naftifine are available as creams for the topical treatment of superficial mycoses. These are usually available over the counter, and there is little to choose between them.
No single antifungal agent is ideal
The main uses and adverse effects of antifungals are summarized in Table 33.8. Although there are several effective preparations available, some conditions such as ringworm infection of the nails or recurrent vaginal candidiasis are frequently intractable to treatment. The number of antifungal agents for systemic fungal infections is limited, and their adverse effects are considerable.
Fungi develop resistance to antifungal agents
Although much less studied than resistance to antimicrobials used against bacteria, there is evidence that many similar mechanisms operate in resistance to antifungals. These include:
• enzyme modification
• target modification
• reduced permeability
• active efflux pumps
• failure to activate antifungal agents.
Resistance involving some or all of these mechanisms has been described in Aspergillus, Candida and Cryptococcus, particularly in the case of the azole compounds.
There is an urgent need for safer more efficacious antifungal agents
Invasive fungal infections are a significant cause of morbidity and mortality in patients undergoing chemotherapy, immune suppression and transplantation. The incidence of these infections is increasing in parallel with the increasing numbers of such patients and their improved survival due to effective antibacterial therapy. New agents to control these infections (e.g. Aspergillus) are needed.
Antiparasitic agents
Parasites pose particular problems
Any consideration of antiparasitic agents must take into account the very large number of different parasites capable of infecting humans, the complexities of their life cycles and the differences between them in their metabolic pathways. Thus, drugs acting against protozoa are usually inactive against helminths and vice versa. Additionally, protozoa and helminths are eukaryotes and therefore metabolically more similar to humans than are bacteria. Although some antibacterials do have antiprotozoal activity (e.g. metronidazole, tetracycline), in general antibacterials are ineffective against parasites. A major challenge has been to identify targets where there are sufficient differences between host and parasite to facilitate safe drug activity. Some of these targets include:
• unique drug uptake: chloroquine, mefloquine, primaquine in malaria
• differences in folic acid metabolism: pyrimethamine in malaria, sulphonamides in toxoplasmosis, trimethoprim in cyclosporiasis
• polyamine uptake: pentamidine in leishmaniasis
• unique trypanothione-dependent reduction mechanisms: fluoromethylornithine against trypanosomes
• unique neurotransmitters: piperazine, ivermectin, pyrantel against nematodes
• cytoskeletal proteins (tubulin): benzimidazoles against nematodes
• intracellular calcium levels: praziquantel against flukes and tapeworms
• oxidative phosphorylation: niclosamide against tapeworms.
Despite differences between host and parasite in these targets, it remains true that a number of the more effective antiparasite drugs carry the risk of significant toxicity.
The wide array of different drugs that have been developed is summarized in Tables 33.9 and 33.10.
Table 33.9 Therapeutic applications of the major antiprotozoal drugs
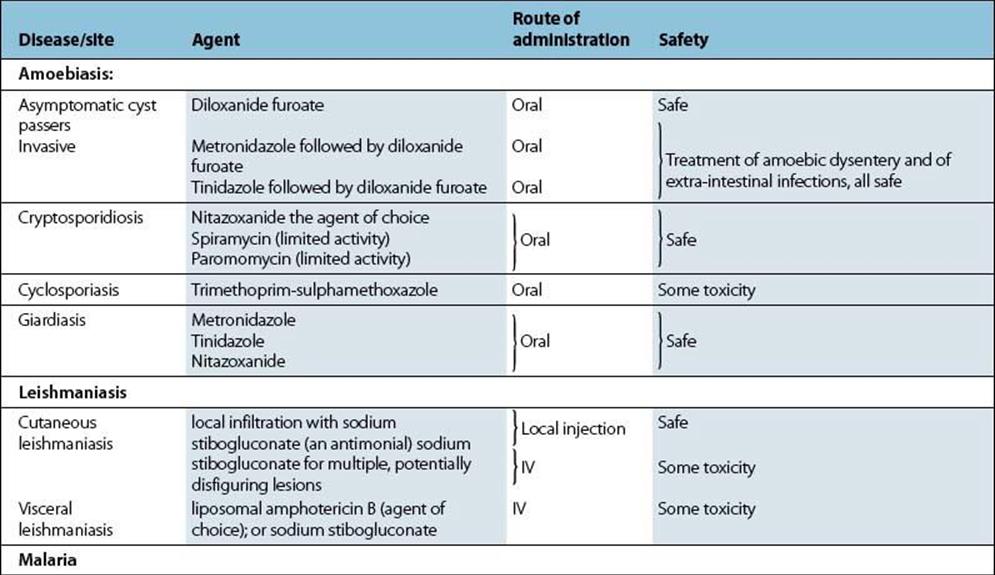
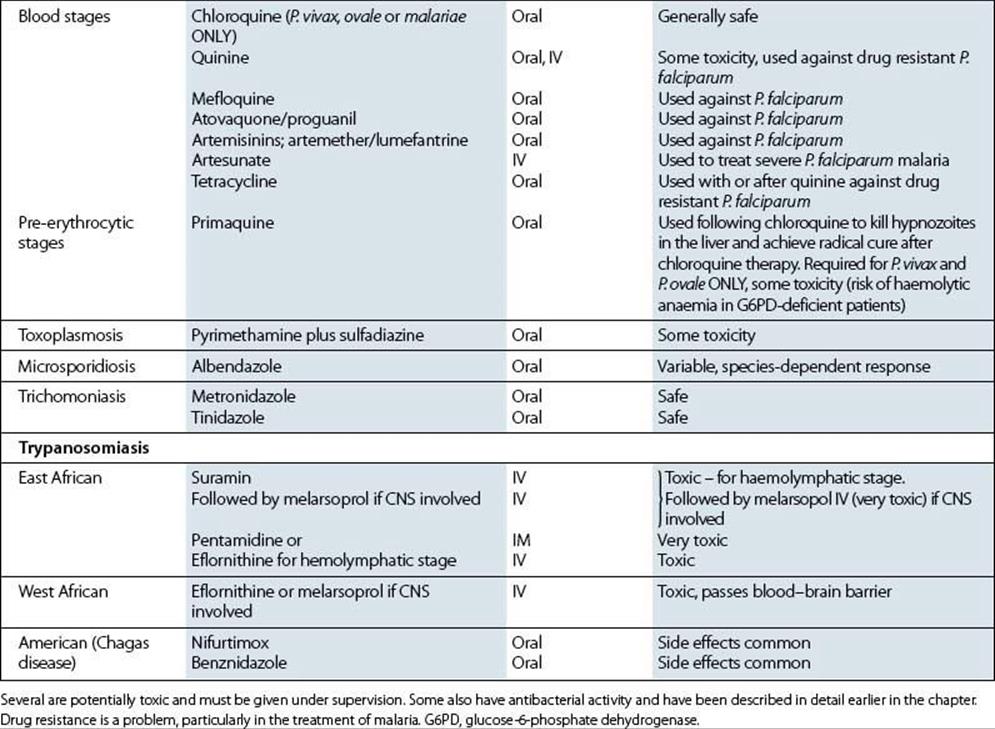
Table 33.10 Therapeutic applications of the major anthelmintic drugs
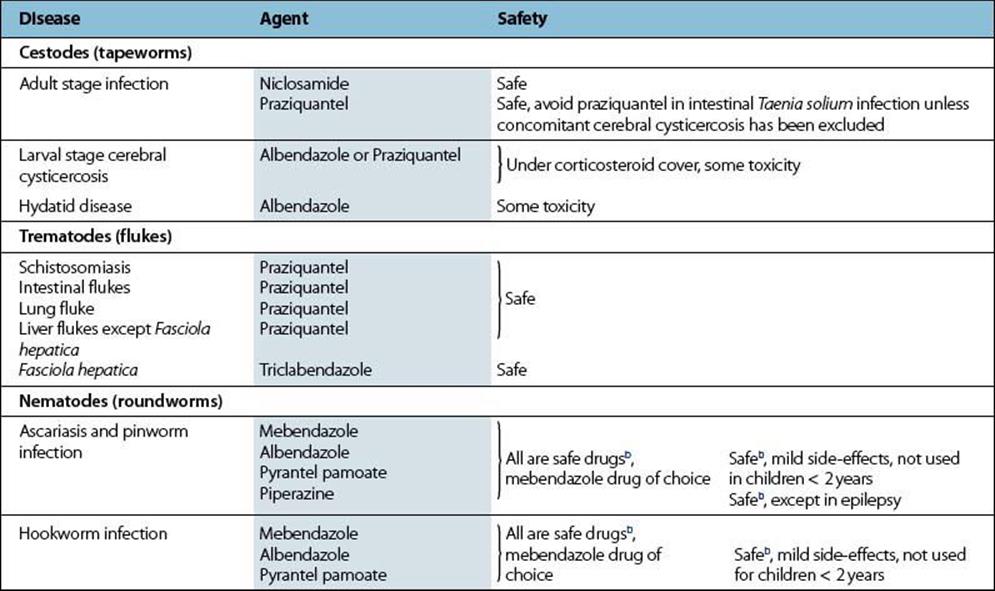
Drug resistance is an increasing problem
As with the antibacterials, drug resistance is a significant problem in the treatment of parasitic infections, particularly with malaria. There are four different indications for antimalarial chemotherapy:
• prophylactic: to prevent infection
• therapeutic: to treat infection (applies to all human malarias)
• radical cure: to prevent relapse following the treatment of acute infection (applies to Plasmodium vivax and P. ovale only)
• killing malarial gametocytes: to prevent transmission.
Falciparum malaria resistant to one or more antimalarial agents is now widespread. Chloroquine-resistant falciparum malaria occurs worldwide and P. vivax also shows focal resistance to this agent, notably in the Asia-Pacific region. The usual alternative to chloroquine in the tropics was combined sulphadoxine/pyrimethamine but there is now significant resistance to the antifolate compounds. Mefloquine-resistant falciparum malaria is found in parts of SE Asia and parts of South America. Quinine, the original antimalarial, is still a first-line agent for severe malaria though it requires careful monitoring during treatment to avoid toxicity. Development of antimalarials from natural products has provided new compounds, the most important being derivatives of artemisinin (from the Chinese drug, quinghaosu, produced from the plant Artemisia annua). Where available, intravenous artesunate has supplanted quinine as the agent of choice for the treatment of severe falciparum malaria. Drug combinations are now deployed for the treatment of falciparum malaria to reduce the chance of developing drug resistance after monotherapy, as happened with chloroquine, and artemisinin combination therapy (ACT) is already replacing quinine. Drug resistance is less of a problem with other protozoa and, although widespread in animal parasitic nematodes, has yet to become a serious issue with human infections.
Protozoa make use of enzyme and target modification to develop resistance (e.g. against antifolates and sulphonamides), but in addition active efflux pumps have been described in resistance of P. falciparum to chloroquine and mefloquine. Resistance to benzimidazole anthelmintics involves target modification, arising from mutations in cuticular tubulins.
Control by chemotherapy versus vaccination
While vaccination is discussed in detail in Chapter 34, it is important to note the role both chemotherapy and vaccination play in protecting individuals. An important difference is that chemotherapy is usually given after exposure to infection, whereas vaccination is usually given before exposure. Chemotherapy essentially offers short-term protection, which wanes once the drug is no longer given; vaccination can give long-term protection without repeated treatment. Vaccination is therefore more effective than chemotherapy in protecting populations.
There are, of course, exceptions to these: passive antibody can be used to treat acute infection just as a drug can, while drugs like pyrimethamine and chloroquine are used for prophylaxis against malaria, almost as if they were short-term vaccines. However, in most cases there is a clear-cut distinction between the one- or two-shot vaccine, conferring protection for years, and the daily or twice-daily drug dose. Naturally, patients and doctors favour the former, whereas the pharmaceutical industry prefers the latter. Therefore, while drug development is carried out by industry without the need for external encouragement, vaccine development needs an outside stimulus in the form of earmarked funding; a field in which the World Health Organization, in particular, has performed with great distinction.
The concept of selectivity, or specificity, is central to both chemotherapy and vaccination
Although they appear so different (Table 33.11), both chemotherapy and vaccination developed together from the intensive study that followed the demonstration in the late 1800s that diseases could be caused by microbes. Pasteur (Box 33.8, Fig. 33.31) showed that killed or weakened microbes (e.g. anthrax, rabies) could be used to induce immunity that was active against that disease, while Ehrlich’s work with histologic dyes led him to the idea that particular chemicals (‘drugs’) might bind specifically to particular microbial structures and damage them, thus being active against several diseases (see Ch. 33). Both therefore established the concept of selectively or specifically targeting infectious organisms within the body as a means of controlling disease.
Table 33.11 Comparison of chemotherapy and vaccination

![]()
Box 33.8 Lessons in Microbiology
Louis Pasteur (1822–1895)
The science of microbiology was established in the nineteenth century by the work of many distinguished scientists. However, one such scientist, Louis Pasteur, may legitimately be regarded as a founding father of this discipline (Fig. 31.9). He, along with Robert Koch, a German doctor (seeCh. 12), was able to show that living organisms or ‘microbes’ were the cause of disease, and provided a firm scientific basis for their study and control.
Pasteur began work at a time when spontaneous generation was still an accepted explanation for the appearance of microorganisms in decaying material. His elegant experiments showed that sterile organic infusions would not putrefy or ferment if there was no subsequent contact with airborne contaminants, proving that spontaneous generation did not occur, and that all microbes must come from pre-existing microbes. This discovery contributed to many fields of science, both basic and applied. Perhaps most important was the contribution Pasteur made to the work of Lister on antiseptics, which revolutionized approaches to surgery.
Pasteur worked in an amazing variety of microbiologic fields, from fermentation in the brewing of beers and production of wines, to identification of silkworm diseases, bringing to each a penetrating scientific insight and making discoveries that brought him national and international renown. His understanding of the roles of organisms in causing diseases, and his acute scientific perception, enabled him to grasp from a series of ‘mishaps’ with experiments on chicken cholera that attenuated microbes could induce not disease, but immunity from disease. His ideas generated powerful opposition, but his belief then was strong enough to encourage him in 1881 to take part in a public trial of his vaccine against anthrax in domestic animals. Later, he used his insight into rabies, caused by organisms he could not see or culture, to develop an attenuated vaccine made from the dried spinal cords of infected rabbits. This was proven effective in humans in 1885, when Pasteur inoculated Joseph Meister, a 9-year-old boy who had been badly bitten by a rabid dog. Meister survived, and Pasteur’s views on vaccination became universally accepted.
Pasteur ended his life as a national hero in his native France, and with a worldwide reputation for his work. His name is immortalized not only in the process of sterilization (‘pasteurization’) that he developed, but in the Institut Pasteur in Paris, which remains one of the most important international centres of microbiologic work.
![]()

Figure 33.31 Louis Pasteur (1822–1895).
The specificity of an antimicrobial drug resides in its ability to damage the microbe and not the host
As noted earlier, antimicrobial drugs should ideally bind to a molecule present only in the microbe to ensure specificity for the microbe and not the host. The extent to which this can be achieved varies from microbe to microbe. Bacteria, with their prokaryotic cell structure, are much more remote from humans than fungi, protozoa or worms (which are all eukaryotic). It is not surprising, therefore, that the most effective antibiotics are generally those used against bacteria. As much of the viral life cycle uses host cell components, antiviral chemotherapy has been much less successful than antibacterial therapy.
Many antimicrobial agents are products of microbes themselves or derivatives of these products. It is presumed that they form part of the self-preservation mechanism by which the microbes prevent overcrowding with their own or other species.
Although it is possible to administer antimicrobial agents in ways that prolong their presence in the body, they are no longer active once concentrations fall below a critical threshold. Continuing antimicrobial activity therefore requires repeated administration as opposed to vaccines which can provide long-term protection with far less re-administration (see Ch. 34).
Control versus eradication
Control and eradication are different objectives, although eradication is always an ideal endpoint
Many infections can be controlled (at least in some parts of the world) by the use of a combination of strategies, including chemotherapy and vaccination (see Ch. 34) (Table 33.11), but are certainly not eradicated, even in those countries where control is most effective. Epidemiologic theory (see Ch. 31) predicts that once transmission rates fall below a threshold value, the infection should die out, and this may certainly be true at a local level. However, reservoirs of infection persist where treatment is non-existent or ineffective, or infection is re-introduced by the movement of peoples, and new epidemics may therefore develop. To date, only one disease – smallpox – has been taken to the point at which the organism has been eliminated. What are the chances that other infectious diseases will follow smallpox into oblivion? Various factors are important in determining the effectiveness of any eradication programme (Table 33.12).
Table 33.12 Strategies for control of infectious diseases
|
General features |
Water purification (water-borne diseases) |
|
Food |
Cold storage |
|
Zoonoses and arthropod-transmitted infections |
Control of vectors (mosquitoes, ticks, lice, etc.) |
|
Specific disease treatment or prevention |
Chemotherapy |
|
Miscellaneous measures |
Changes in personal habits (reduced promiscuity, use of condoms, improved personal hygiene, etc.) |
Realism is required when considering the long-term aims of antimicrobial control strategies
Hopes raised by the early success of antibiotics were soon dashed by the emergence of resistance, and far from the microbial load borne by the human race being diminished in recent years, it has if anything increased. Many infections covered in this book, HIV, Legionnaires’ disease, Lyme disease, to name but a few, do not feature in older textbooks of microbiology. Infections previously well controlled by antibiotics have become serious problems in hospitals (MRSA, C. difficile). Approaches to the control of infectious diseases are therefore a matter of identifying priorities such as:
• Which diseases could, with suitable effort, be eradicated?
• Would the cost of eradication be justified?
• Which diseases need urgent measures to stop them getting worse?
• Which diseases are responsible for the most human suffering and economic loss?
Inevitably, some diseases will not feature strongly on any such list, and it must be accepted that they may always be with us.
Use and misuse of antimicrobial agents
Much has been said in this chapter about the interactions between antimicrobial agents and microbes – the mechanisms of selective toxicity and the defences put up by resistant organisms. The distribution, metabolism and excretion of agents by the host have been considered briefly, together with the important toxic side effects of the agents. The choice of antimicrobial for treating specific infections is dealt with in the appropriate systems chapter (Chs 18–30). Dosage regimens have not been included because they vary with the agent, the infection, the age and the underlying condition of the patient, and sometimes from one country to another. Practitioners should consult appropriate local pharmacy guidelines.
Antimicrobial agents should only be used appropriately for prophylaxis or treatment
In conclusion, we should stand back and ask ‘Is antimicrobial therapy necessary for this patient, and, if so, which agent is appropriate?’ Antimicrobial agents can be used:
• to help prevent infection (prophylaxis)
• to treat infection.
Prophylactic use of antibiotics is appropriate only in a few clearly defined circumstances and is usually of limited duration (e.g. 1–2 days). Specific examples include: (1) patients of normal susceptibility who have been exposed to specific pathogens (e.g. bacterial meningitis or tuberculosis), (2) individuals with increased susceptibility to infection (e.g. neutropenic patients), and (3) perioperative antibiotic ‘cover’ for patients undergoing surgery.
Antimicrobial use results in the selection of resistant strains
If antibiotic treatment is necessary, several factors must be considered, and these are summarized in Figure 33.32. It is important to recognize that during treatment not only the infecting microbe, but also the patient and all his or her normal microbial flora are being exposed to the effects of the antimicrobial agent. Use of antimicrobials has been clearly shown to select for resistant strains, both in the individual and in the community, and overuse or inappropriate use only increases this risk. History suggests that microbes will never run out of ways of developing resistance, but we may run out of effective antimicrobials.
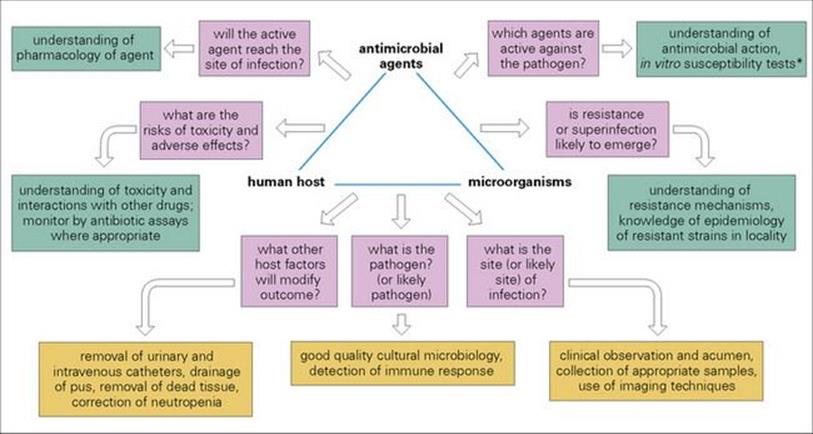
Figure 33.32 The interactions between antimicrobial agents, microorganisms and the human host can be summarized by examining the answers to several questions affecting each side of the triangle of interaction. * Other tests include phenotypic and genotypic antiviral susceptibility tests and viral load tests.
![]()
 Key Facts
Key Facts
• Infection is unique among the diseases which afflict mankind because it involves two distinct biological systems. Antimicrobial agents are designed to inhibit one system (the microbe) while doing minimal damage to the other (the patient). Antimicrobial agents require selective toxicity.
• Antimicrobial agents are often themselves products of microorganisms (natural products) although most are chemically modified to improve their properties. Other agents are entirely synthetic. Antibacterials are the most numerous; designing antiviral, antifungal and antiparasitic drugs which are selectively toxic provides much greater challenges.
• Antibacterials are classified by their target site and their chemical family; this helps us to understand better their mode of action and the mechanisms of resistance.
• Antibacterials have four possible sites of action in the bacterial cell: cell wall, protein, nucleic acids and cell membrane. The majority act at the cell wall or inhibit protein or nucleic acid synthesis. At each site there are many different molecular targets (enzymes or substrates) which can be specifically inhibited.
• Development of resistance is the major limiting factor of antibacterials. It arises through random mutation of bacterial chromosomal genes but more importantly through acquisition, from other bacteria, of resistance genes on integrons, transposons and plasmids.
• Mutated or acquired genes confer resistance by altering the target site of the antibacterial, altering the uptake of the drug, or producing drug-destroying enzymes.
• The emergence of AIDS has provided an enormous stimulus to research in antivirals (especially anti-HIV drugs). Selective toxicity is again a major challenge. Drug combinations show promise in the treatment of HIV, but there is no specific therapy for the majority of viral diseases. Effective therapy is available for other viral infections, including hepatitis B and C, influenza A and B, HSV and CMV.
• The number of classes of antifungal molecules is very limited. Toxicity (all), difficulty of formulation (polyenes), and emerging resistance (azoles) make effective treatment of fungal infections a serious challenge.
• Although there are many antiparasitic drugs available, a number show toxicity and others are becoming increasingly ineffective because of the development of resistance. This is particularly so in malaria infections, where parasites show resistance to almost all drugs presently available.
• Bacteria can be tested in the laboratory for their susceptibility to antibacterials. The results of well-controlled tests provide a valuable guide to appropriate treatment. In vitro tests with antifungals are less reliable and are rarely performed with antivirals in the clinical laboratory setting.
![]()