Harper’s Illustrated Biochemistry, 29th Edition (2012)
SECTION II. Bioenergetics & the Metabolism of Carbohydrates & Lipids
Chapter 25. Lipid Transport & Storage
Kathleen M. Botham, PhD, DSc & Peter A. Mayes, PhD, DSc
OBJECTIVES
After studying this chapter, you should be able to:
![]() Identify the four major groups of plasma lipoproteins and the four major lipid classes they carry.
Identify the four major groups of plasma lipoproteins and the four major lipid classes they carry.
![]() Illustrate the structure of a lipoprotein particle.
Illustrate the structure of a lipoprotein particle.
![]() Indicate the major types of apolipoprotein found in the different lipoprotein classes.
Indicate the major types of apolipoprotein found in the different lipoprotein classes.
![]() Explain that triacylglycerol is carried from the intestine (after intake from the diet) to the liver in chylomicrons and from the liver to extrahepatic tissues in very low density lipoprotein (VLDL), and these particles are synthesized in intestinal and liver cells, respectively, by similar processes.
Explain that triacylglycerol is carried from the intestine (after intake from the diet) to the liver in chylomicrons and from the liver to extrahepatic tissues in very low density lipoprotein (VLDL), and these particles are synthesized in intestinal and liver cells, respectively, by similar processes.
![]() Illustrate the processes by which chylomicrons are metabolized by lipases to form chylomicron remnants, which are then removed from the circulation by the liver.
Illustrate the processes by which chylomicrons are metabolized by lipases to form chylomicron remnants, which are then removed from the circulation by the liver.
![]() Explain how VLDL is metabolized by lipases to VLDL remnants (also called intermediate-density lipoprotein (IDL)) which may be cleared by the liver or converted to low-density lipoprotein (LDL), which functions to deliver cholesterol from the liver to extrahepatic tissues and is taken up via the LDL (apoB100,E) receptor.
Explain how VLDL is metabolized by lipases to VLDL remnants (also called intermediate-density lipoprotein (IDL)) which may be cleared by the liver or converted to low-density lipoprotein (LDL), which functions to deliver cholesterol from the liver to extrahepatic tissues and is taken up via the LDL (apoB100,E) receptor.
![]() Explain how high-density lipoprotein (HDL), which returns cholesterol from extrahepatic tissues to the liver in reverse cholesterol transport, is synthesized, indicate the mechanisms by which it accepts cholesterol from tissues, and show how it is metabolized in the HDL cycle.
Explain how high-density lipoprotein (HDL), which returns cholesterol from extrahepatic tissues to the liver in reverse cholesterol transport, is synthesized, indicate the mechanisms by which it accepts cholesterol from tissues, and show how it is metabolized in the HDL cycle.
![]() Understand how the liver plays a central role in lipid transport and metabolism and how hepatic VLDL secretion is regulated by the diet and hormones.
Understand how the liver plays a central role in lipid transport and metabolism and how hepatic VLDL secretion is regulated by the diet and hormones.
![]() Be aware of the roles of LDL and HDL in promoting and retarding, respectively, the development of atherosclerosis.
Be aware of the roles of LDL and HDL in promoting and retarding, respectively, the development of atherosclerosis.
![]() Indicate the causes of alcoholic and nonalcoholic fatty liver disease.
Indicate the causes of alcoholic and nonalcoholic fatty liver disease.
![]() Appreciate that adipose tissue is the main store of triacylglycerol in the body and explain the processes by which fatty acids are released and how they are regulated.
Appreciate that adipose tissue is the main store of triacylglycerol in the body and explain the processes by which fatty acids are released and how they are regulated.
![]() Understand the role of brown adipose tissue in the generation of body heat.
Understand the role of brown adipose tissue in the generation of body heat.
BIOMEDICAL IMPORTANCE
Fat absorbed from the diet and lipids synthesized by the liver and adipose tissue must be transported between the various tissues and organs for utilization and storage. Since lipids are insoluble in water, the problem of how to transport them in the aqueous blood plasma is solved by associating nonpolar lipids (triacylglycerol and cholesteryl esters) with amphipathic lipids (phospholipids and cholesterol) and proteins to make water-miscible lipoproteins.
In a meal-eating omnivore such as the human, excess calories are ingested in the anabolic phase of the feeding cycle, followed by a period of negative caloric balance when the organism draws upon its carbohydrate and fat stores. Lipoproteins mediate this cycle by transporting lipids from the intestines as chylomicrons—and from the liver as very low density lipoproteins (VLDL)—to most tissues for oxidation and to adipose tissue for storage. Lipid is mobilized from adipose tissue as free fatty acids (FFA) bound to serum albumin. Abnormalities of lipoprotein metabolism cause various hypo- or hyperlipoproteinemias. The most common of these is in diabetes mellitus, where insulin deficiency causes excessive mobilization of FFA and underutilization of chylomicrons and VLDL, leading to hypertriacylglycerolemia. Most other pathologic conditions affecting lipid transport are due primarily to inherited defects, some of which cause hypercholesterolemia and premature atherosclerosis (Table 26-1). Obesity—particularly abdominal obesity—is a risk factor for increased mortality, hypertension, type 2 diabetes mellitus, hyperlipidemia, hyperglycemia, and various endocrine dysfunctions.
LIPIDS ARE TRANSPORTED IN THE PLASMA AS LIPOPROTEINS
Four Major Lipid Classes Are Present in Lipoproteins
Plasma lipids consist of triacylglycerols (16%), phospholipids (30%), cholesterol (14%), and cholesteryl esters (36%) and a much smaller fraction of unesterified long-chain fatty acids (free fatty acids or FFA) (4%). This latter fraction, the FFA, is metabolically the most active of the plasma lipids.
Four Major Groups of Plasma Lipoproteins Have Been Identified
Since fat is less dense than water, the density of a lipoprotein decreases as the proportion of lipid to protein increases (Table 25-1). Four major groups of lipoproteins have been identified that are important physiologically and in clinical diagnosis. These are (1) chylomicrons, derived from intestinal absorption of triacylglycerol and other lipids; (2) very low density lipoproteins (VLDL, or pre-β-lipoproteins), derived from the liver for the export of triacylglycerol; (3) low-density lipoproteins (LDL, or β-lipoproteins), representing a final stage in the catabolism of VLDL; and (4) high-density lipoproteins (HDL, or α-lipoproteins), involved in cholesterol transport and also in VLDL and chylomicron metabolism. Triacylglycerol is the predominant lipid in chylomicrons and VLDL, whereas cholesterol and phospholipid are the predominant lipids in LDL and HDL, respectively (Table 25-1). Lipo-proteins may be separated according to their electrophoretic properties into α-, β-, and pre-β-lipoproteins.
TABLE 25–1 Composition of the Lipoproteins in Plasma of Humans
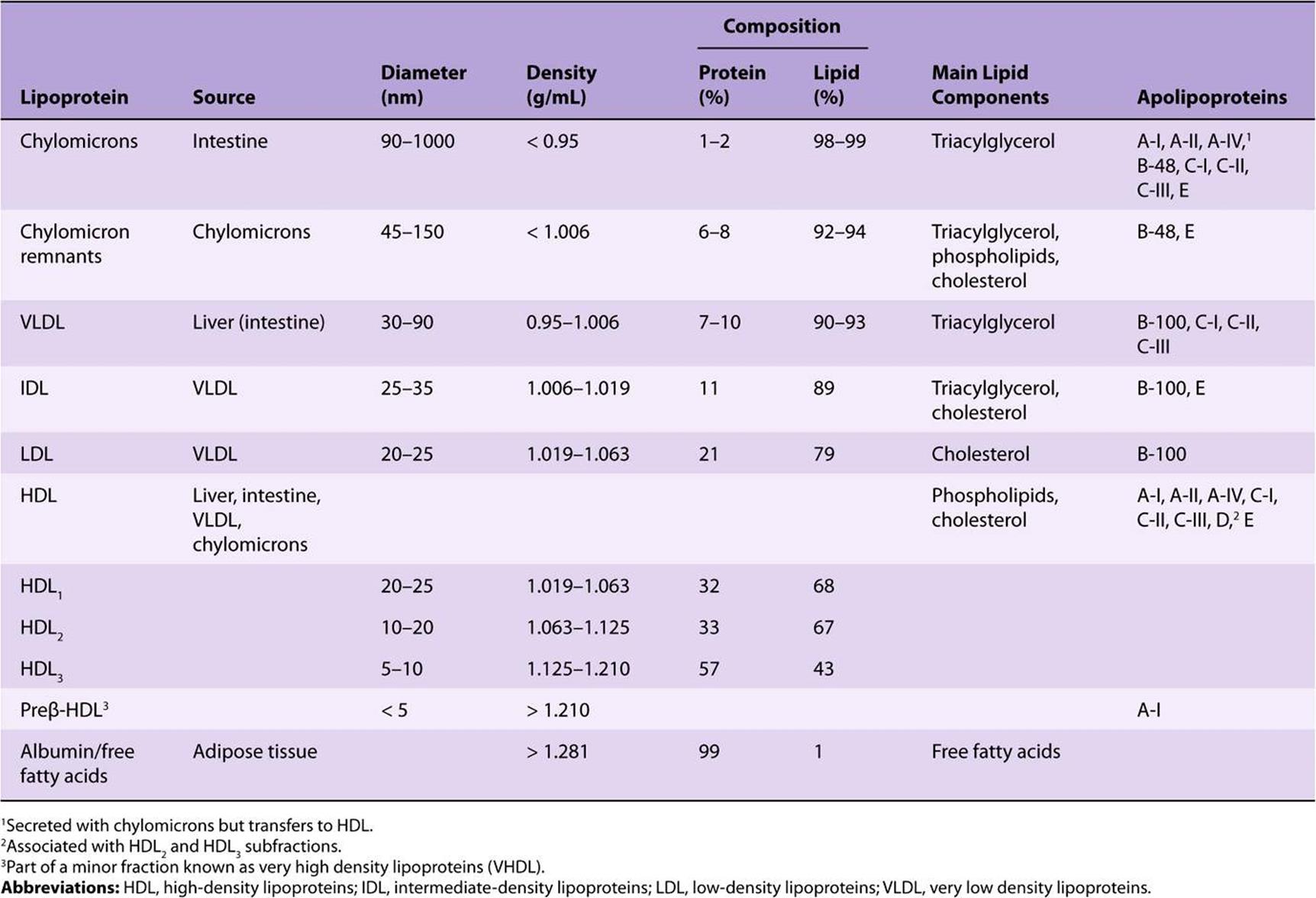
Lipoproteins Consist of a Nonpolar Core & a Single Surface Layer of Amphipathic Lipids
The nonpolar lipid core consists of mainly triacylglycerol and cholesteryl ester and is surrounded by a single surface layer of amphipathic phospholipid and cholesterol molecules (Figure 25–1). These are oriented so that their polar groups face outward to the aqueous medium, as in the cell membrane (Chapter 15). The protein moiety of a lipoprotein is known as an apolipoprotein or apoprotein, constituting nearly 70% of some HDL and as little as 1% of chylomicrons. Some apolipoproteins are integral and cannot be removed, whereas others are free to transfer to other lipoproteins.
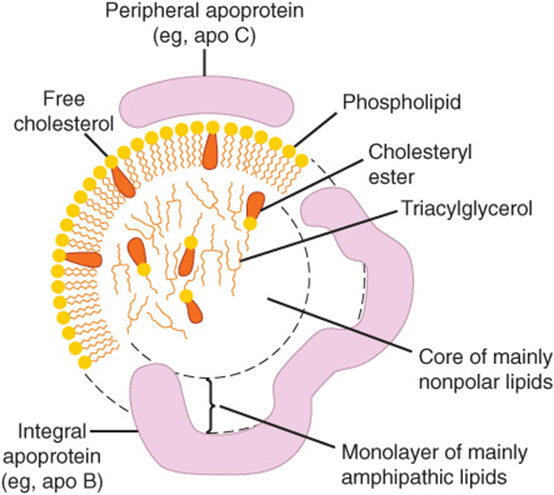
FIGURE 25–1 Generalized structure of a plasma lipoprotein. The similarities with the structure of the plasma membrane are to be noted. Small amounts of cholesteryl ester and triacylglycerol are found in the surface layer and a little free cholesterol in the core.
The Distribution of Apolipoproteins Characterizes the Lipoprotein
One or more apolipoproteins (proteins or polypeptides) are present in each lipoprotein. The major apolipoproteins of HDL (α-lipoprotein) are designated A (Table 25-1). The main apolipoprotein of LDL (β-lipoprotein) is apolipoprotein B (B-100), which is found also in VLDL. Chylomicrons contain a truncated form of apo B (B-48) that is synthesized in the intestine, while B-100 is synthesized in the liver. Apo B-100 is one of the longest single polypeptide chains known, having 4536 amino acids and a molecular mass of 550,000 Da. Apo B-48 (48% of B-100) is formed after transcription of the apo B-100 gene by the introduction of a stop signal into the mRNA transcript by an RNA editing enzyme. Apo C-I, C-II, and C-III are smaller polypeptides (molecular mass 7000-9000 Da) freely transferable between several different lipoproteins. Apo E, found in VLDL, HDL, chylomicrons, and chylomicron remnants, is also freely transferable; it accounts for 5-10% of total VLDL apolipoproteins in normal subjects.
Apolipoproteins carry out several roles: (1) they can form part of the structure of the lipoprotein, eg, apo B; (2) they are enzyme cofactors, eg, C-II for lipoprotein lipase, A-I for lecithin:cholesterol acyltransferase, or enzyme inhibitors, eg, apo A-II and apo C-III for lipoprotein lipase, apo C-I for cholesteryl ester transfer protein; and (3) they act as ligands for interaction with lipoprotein receptors in tissues, eg, apo B-100 and apo E for the LDL receptor, apo E for the LDL-receptor-related protein (LRP), which has been identified as the remnant receptor, and apo A-I for the HDL receptor. The functions of apo A-IV and apo D, however, are not yet clearly defined, although apo D is believed to be an important factor in human neurodegenerative disorders.
FREE FATTY ACIDS ARE RAPIDLY METABOLIZED
The FFA (also termed nonesterified fatty acids or unesterified fatty acids) arise in the plasma from the breakdown of triacylglycerol in adipose tissue or as a result of the action of lipoprotein lipase on the plasma triacylglycerols. They are found in combination with albumin, a very effective solubilizer, in concentrations varying between 0.1 and 2.0 μeq/mL of plasma. Levels are low in the fully fed condition and rise to 0.7-0.8 μeq/mL in the starved state. In uncontrolled diabetes mellitus, the level may rise to as much as 2 μeq/mL.
FFA are removed from the blood extremely rapidly and oxidized (fulfilling 25-50% of energy requirements in starvation) or esterified to form triacylglycerol in the tissues. In starvation, esterified lipids from the circulation or in the tissues are oxidized as well, particularly in heart and skeletal muscle cells, where considerable stores of lipid are to be found.
The FFA uptake by tissues is related directly to the plasma-FFA concentration, which in turn is determined by the rate of lipolysis in adipose tissue. After dissociation of the fatty acid-albumin complex at the plasma membrane, fatty acids bind to a membrane fatty acid transport protein that acts as a transmembrane cotransporter with Na+. On entering the cytosol, FFA are bound by intracellular fatty-acid-binding proteins. The role of these proteins in intracellular transport is thought to be similar to that of serum albumin in extracellular transport of long-chain fatty acids.
TRIACYLGLYCEROL IS TRANSPORTED FROM THE INTESTINES IN CHYLOMICRONS & FROM THE LIVER IN VERY LOW DENSITY LIPOPROTEINS
By definition, chylomicrons are found in chyle formed only by the lymphatic system draining the intestine. They are responsible for the transport of all dietary lipids into the circulation. Small quantities of VLDL are also to be found in chyle; however, most VLDL in the plasma are of hepatic origin. They are the vehicles of transport of triacylglycerol from the liver to the extrahepatic tissues.
There are striking similarities in the mechanisms of formation of chylomicrons by intestinal cells and of VLDL by hepatic parenchymal cells (Figure 25–2), perhaps because—apart from the mammary gland—the intestine and liver are the only tissues from which particulate lipid is secreted. Newly secreted or “nascent” chylomicrons and VLDL contain only a small amount of apolipoproteins C and E, and the full complement is acquired from HDL in the circulation (Figures 25-3 and 25-4). Apo B, however, is an integral part of the lipoprotein particles, it is incorporated inside the cells and is essential for chylomicron and VLDL formation. In abetalipoproteinemia (a rare disease), lipoproteins containing apo B are not formed and lipid droplets accumulate in the intestine and liver.
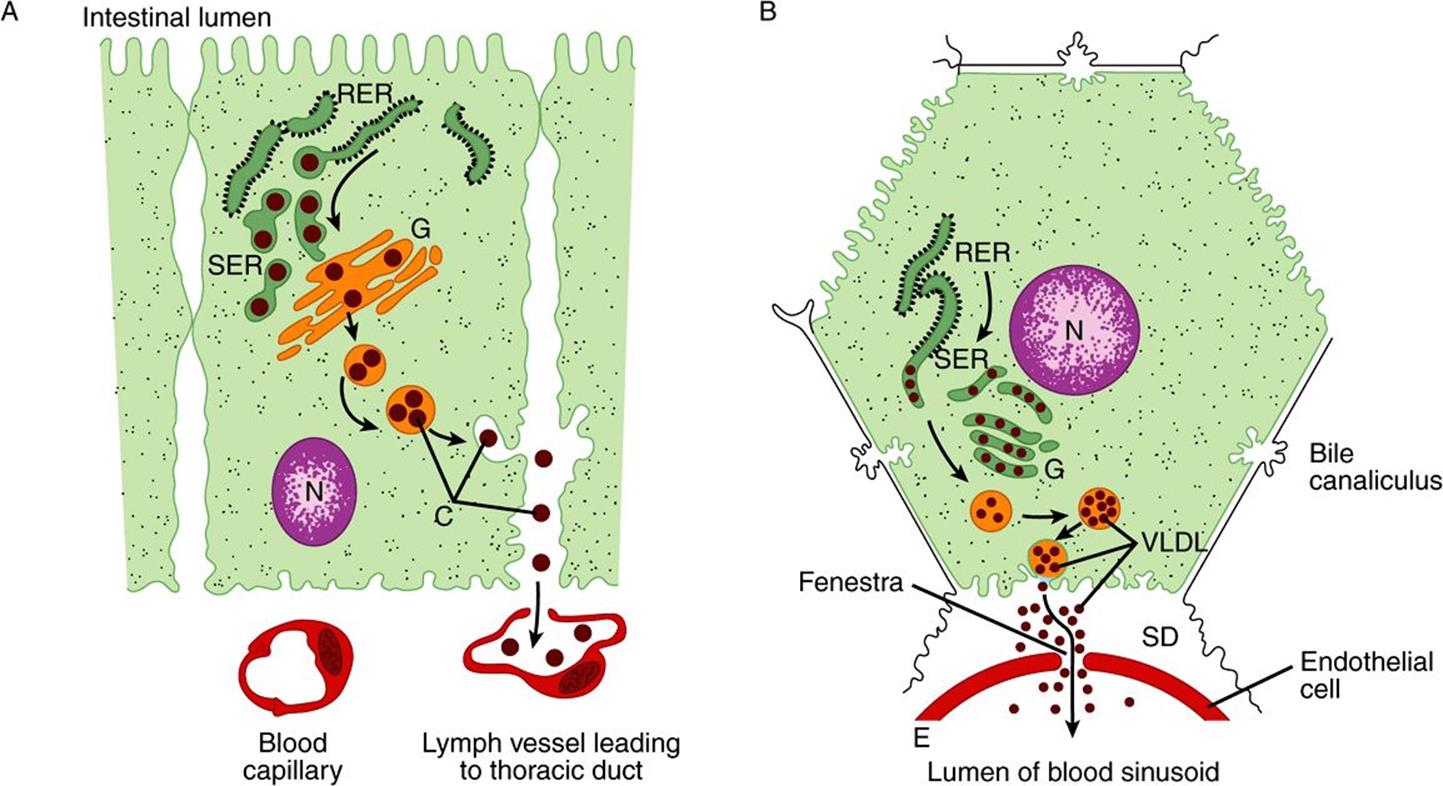
FIGURE 25–2 The formation and secretion of (A) chylomicrons by an intestinal cell and (B) very low density lipoproteins by a hepatic cell. (C, chylomicrons; E, endothelium; G, Golgi apparatus; N, nucleus; RER, rough endoplasmic reticulum; SD, space of Disse, containing blood plasma; SER, smooth endoplasmic reticulum; VLDL, very low density lipoproteins.) Apolipoprotein B, synthesized in the RER, is incorporated into particles with triacylglycerol, cholesterol, and phospholipids in the SER. After the addition of carbohydrate residues in G, they are released from the cell by reverse pinocytosis. Chylomicrons pass into the lymphatic system. VLDL are secreted into the space of Disse and then into the hepatic sinusoids through fenestrae in the endothelial lining.
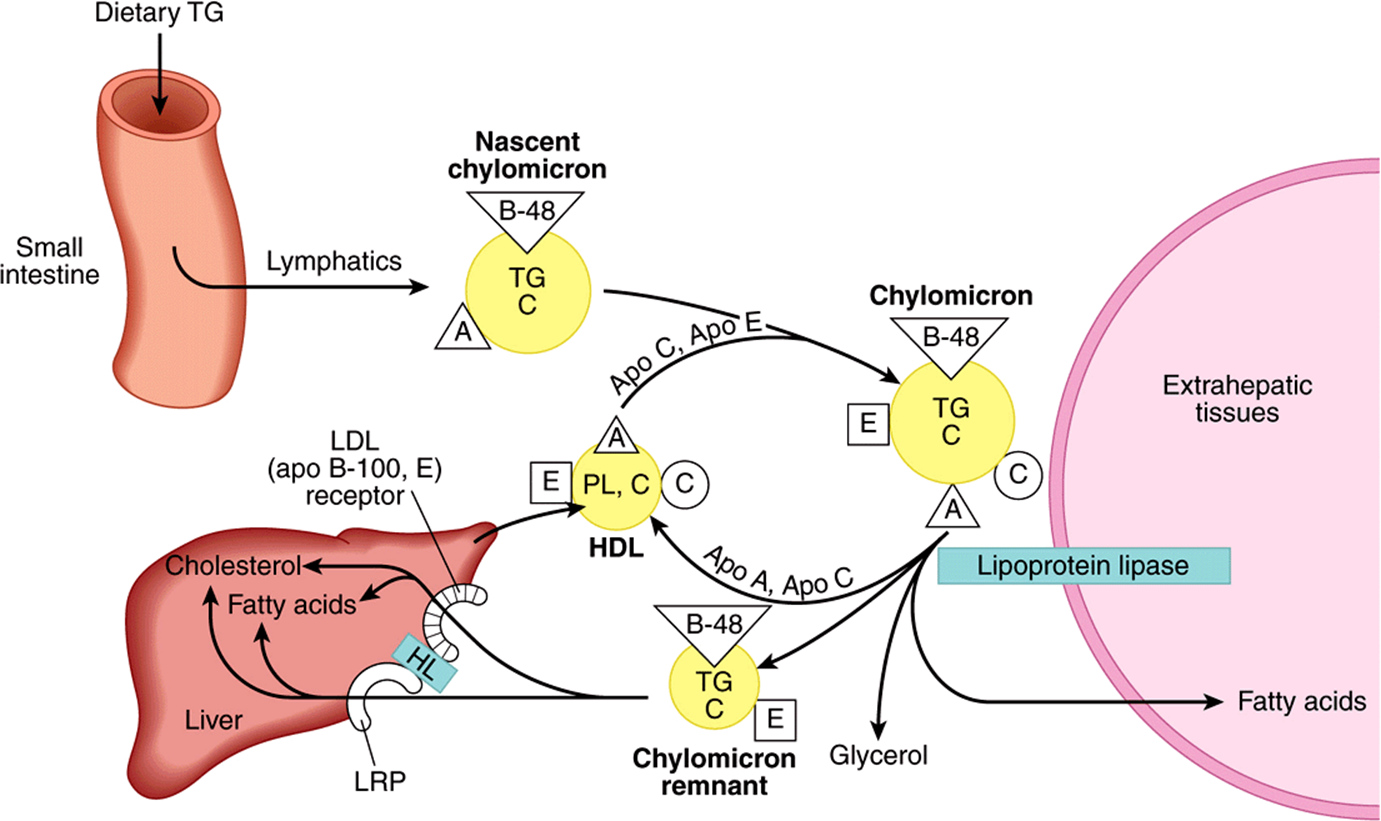
FIGURE 25–3 Metabolic fate of chylomicrons. (A, apolipoprotein A; B-48, apolipoprotein B-48; C, apolipoprotein C; C, cholesterol and cholesteryl ester; E, apolipoprotein E; HDL, high-density lipoprotein; HL, hepatic lipase; LRP, LDL-receptor-related protein; PL, phospholipid; TG, triacylglycerol.) Only the predominant lipids are shown.
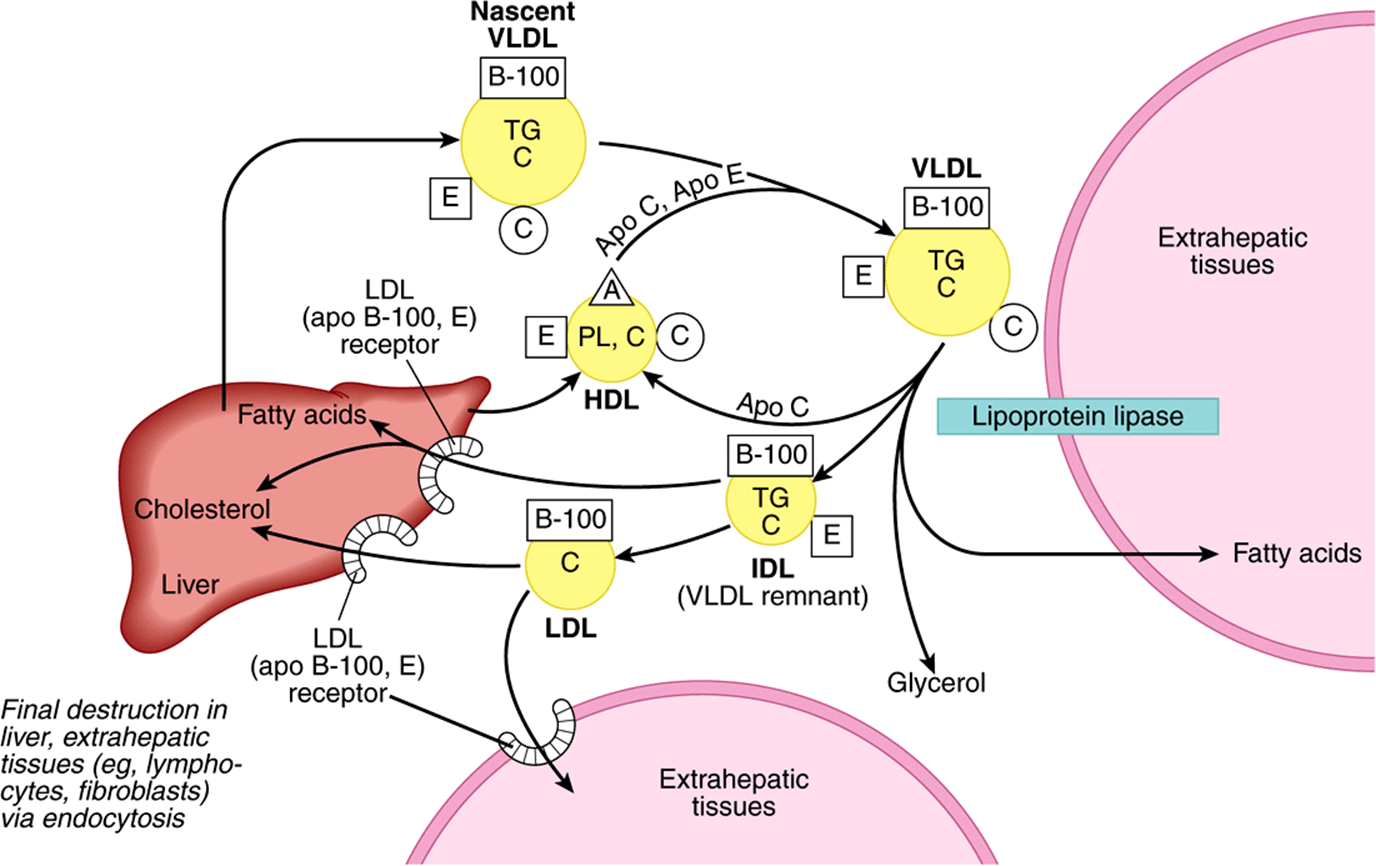
FIGURE 25–4 Metabolic fate of very low density lipoproteins (VLDL) and production of low-density lipoproteins (LDL). (A, apolipoprotein A; B-100, apolipoprotein B-100; C, apolipoprotein C; C, cholesterol and cholesteryl ester; E, apolipoprotein E; HDL, high-density lipoprotein; IDL, intermediate-density lipoprotein; PL, phospholipid; TG, triacylglycerol.) Only the predominant lipids are shown. It is possible that some IDL is also metabolized via the low density lipoproten receptor-related protein (LRP.)
A more detailed account of the factors controlling hepatic VLDL secretion is given below.
CHYLOMICRONS & VERY LOW DENSITY LIPOPROTEINS ARE RAPIDLY CATABOLIZED
The clearance of chylomicrons from the blood is rapid, the half-time of disappearance being under 1 h in humans. Larger particles are catabolized more quickly than smaller ones. Fatty acids originating from chylomicron triacylglycerol are delivered mainly to adipose tissue, heart, and muscle (80%), while ~20% goes to the liver. However, the liver does not metabolize native chylomicrons or VLDL significantly; thus, the fatty acids in the liver must be secondary to their metabolism in extrahepatic tissues.
Triacylglycerols of Chylomicrons & VLDL Are Hydrolyzed by Lipoprotein Lipase
Lipoprotein lipase is located on the walls of blood capillaries, anchored to the endothelium by negatively charged proteoglycan chains of heparan sulfate. It has been found in heart, adipose tissue, spleen, lung, renal medulla, aorta, diaphragm, and lactating mammary gland, although it is not active in adult liver. It is not normally found in blood; however, following injection of heparin, lipoprotein lipase is released from its heparan sulfate binding sites into the circulation. Hepatic lipase is bound to the sinusoidal surface of liver cells and is also released by heparin. This enzyme, however, does not react readily with chylomicrons or VLDL but is involved in chylomicron remnant and HDL metabolism.
Both phospholipids and apo C-II are required as cofactors for lipoprotein lipase activity, while apo A-II and apo C-III act as inhibitors. Hydrolysis takes place while the lipoproteins are attached to the enzyme on the endothelium. Triacylglycerol is hydrolyzed progressively through a diacylglycerol to a monoacylglycerol and finally to FFA plus glycerol. Some of the released FFA return to the circulation, attached to albumin, but the bulk is transported into the tissue (Figures 25-3 and 25-4). Heart lipoprotein lipase has a low K for triacylglycerol, about one-tenth of that for the enzyme in adipose tissue. This enables the delivery of fatty acids from triacylglycerol to be redirected from adipose tissue to the heart in the starved state when the plasma triacylglycerol decreases. A similar redirection to the mammary gland occurs during lactation, allowing uptake of lipoprotein triacylglycerol fatty acid for milk fat synthesis. The VLDL receptor plays an important part in the delivery of fatty acids from VLDL triacylglycerol to adipocytes by binding VLDL and bringing it into close contact with lipoprotein lipase. In adipose tissue, insulin enhances lipoprotein lipase synthesis in adipocytes and its translocation to the luminal surface of the capillary endothelium.
The Action of Lipoprotein Lipase Forms Remnant Lipoproteins
Reaction with lipoprotein lipase results in the loss of 70-90% of the triacylglycerol of chylomicrons and in the loss of apo C (which returns to HDL) but not apo E, which is retained. The resulting chylomicron remnant is about half the diameter of the parent chylomicron and is relatively enriched in cholesterol and cholesteryl esters because of the loss of triacylglycerol (Figure 25–3). Similar changes occur to VLDL, with the formation of VLDL remnants(also called intermediatedensity lipoprotein (IDL) (Figure 25–4).
The Liver Is Responsible for the Uptake of Remnant Lipoproteins
Chylomicron remnants are taken up by the liver by receptormediated endocytosis, and the cholesteryl esters and triacylglycerols are hydrolyzed and metabolized. Uptake is mediated by apo E (Figure 25–3), via two apo E-dependent receptors, the LDL (apo B-100, E) receptor and the LRP (LDL receptor-related protein). Hepatic lipase has a dual role: (1) it acts as a ligand to facilitate remnant uptake and (2) it hydrolyzes remnant triacylglycerol and phospholipid.
After metabolism to IDL, VLDL may be taken up by the liver directly via the LDL (apo B-100, E) receptor, or it may be converted to LDL. Only one molecule of apo B-100 is present in each of these lipoprotein particles, and this is conserved during the transformations. Thus, each LDL particle is derived from a single precursor VLDL particle (Figure 25–4). In humans, a relatively large proportion of IDL forms LDL, accounting for the increased concentrations of LDL in humans compared with many other mammals.
LDL IS METABOLIZED VIA THE LDL RECEPTOR
The liver and many extrahepatic tissues express the LDL (apo B-100, E) receptor. It is so designated because it is specific for apo B-100 but not B-48, which lacks the carboxyl terminal domain of B-100 containing the LDL receptor ligand, and it also takes up lipoproteins rich in apo E. Approximately 30% of LDL is degraded in extrahepatic tissues and 70% in the liver. A positive correlation exists between the incidence of atherosclerosis and the plasma concentration of LDL cholesterol. The LDL (apoB-100, E) receptor is defective in familial hypercholesterolemia, a genetic condition which blood LDL cholesterol levels are increased, causing premature atherosclerosis (Table 26-1). For further discussion of the regulation of the LDL receptor, see Chapter 26.
HDL TAKES PART IN BOTH LIPOPROTEIN TRIACYLGLYCEROL & CHOLESTEROL METABOLISM
HDL is synthesized and secreted from both liver and intestine (Figure 25–5). However, apo C and apo E are synthesized in the liver and transferred from liver HDL to intestinal HDL when the latter enters the plasma. A major function of HDL is to act as a repository for the apo C and apo E required in the metabolism of chylomicrons and VLDL. Nascent HDL consists of discoid phospholipid bilayers containing apo A and free cholesterol. These lipoproteins are similar to the particles found in the plasma of patients with a deficiency of the plasma enzyme lecithin:cholesterol acyltransferase (LCAT) and in the plasma of patients with obstructive jaundice. LCAT—and the LCAT activator apo A-I—bind to the discoidal particles, and the surface phospholipid and free cholesterol are converted into cholesteryl esters and lysolecithin (Chapter 24). The nonpolar cholesteryl esters move into the hydrophobic interior of the bilayer, whereas lysolecithin is transferred to plasma albumin. Thus, a nonpolar core is generated, forming a spherical, pseudomicellar HDL covered by a surface film of polar lipids and apolipoproteins. This aids the removal of excess unesterified cholesterol from lipoproteins and tissues as described below. The class B scavenger receptor B1 (SR-B1) has been identified as an HDL receptor with a dual role in HDL metabolism.In the liver and in steroidogenic tissues, it binds HDL via apo A-I, and cholesteryl ester is selectively delivered to the cells, although the particle itself, including apo A-I, is not taken up. In the tissues, on the other hand, SR-B1 mediates the acceptance of cholesterol effluxed from the cells by HDL, which then transports it to the liver for excretion via the bile (either as cholesterol or after conversion to bile acids) in the process known as reverse cholesterol transport (Figure 25–5). HDL3, generated from discoidal HDL by the action of LCAT, accepts cholesterol from the tissues via the SR-B1 and the cholesterol is then esterified by LCAT, increasing the size of the particles to form the less dense HDL2. HDL3 is then reformed, either after selective delivery of cholesteryl ester to the liver via the SR-B1 or by hydrolysis of HDL2 phospholipid and triacylglycerol by hepatic lipase and endothelial lipase. This interchange of HDL2 and HDL3 is called the HDL cycle (Figure 25–5). Free apo A-I is released by these processes and forms preβ-HDL after associating with a minimum amount of phospholipid and cholesterol. Surplus apo A-I is destroyed in the kidney. A second important mechanism for reverse cholesterol transport involves the ATP-binding cassette transporters A1 (ABCA1) and G1 (ABCG1). These transporters are members of a family of transporter proteins that couple the hydrolysis of ATP to the binding of a substrate, enabling it to be transported across the membrane. ABCG1 mediates the transport of cholesterol from cells to HDL, while ABCA1 preferentially promotes efflux to poorly lipidated particles such as preβ-HDL or apo A-1, which are then converted to HDL3 via discoidal HDL (Figure 25–5). Preβ-HDL is the most potent form of HDL inducing cholesterol efflux from the tissues.
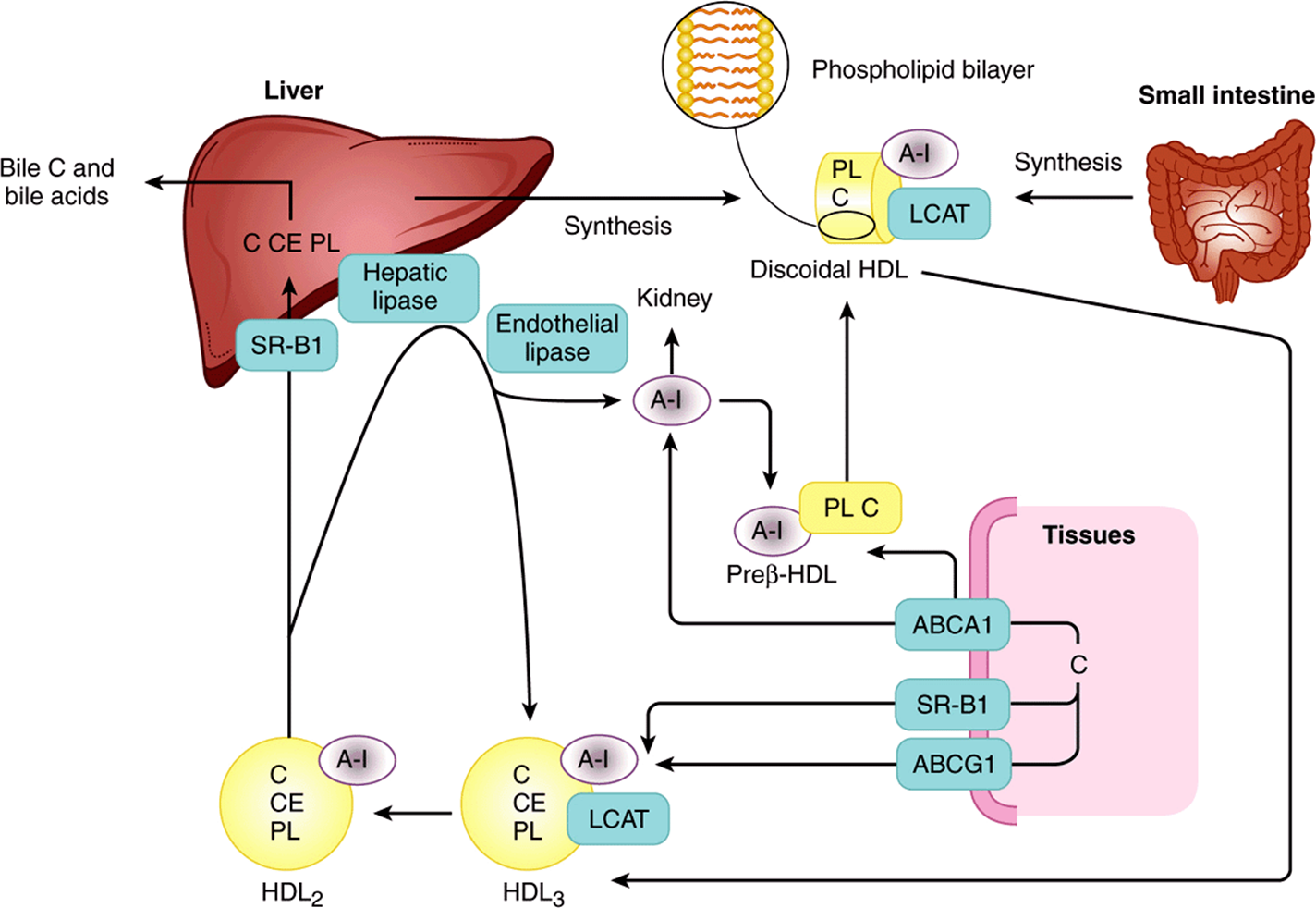
FIGURE 25–5 Metabolism of high-density lipoprotein (HDL) in reverse cholesterol transport. (A-I, apolipoprotein A-I; ABCA 1, ATP-binding cassette transporter A1; ABCG1, ATP-binding cassette transporter G1; C, cholesterol; CE, cholesteryl ester; LCAT, lecithin:cholesterol acyltransferase; PL, phospholipid; SR-B1, scavenger receptor B1.) Preβ-HDL, HDL2, HDL3—see Table 25-1. Surplus surface constituents from the action of lipoprotein lipase on chylomicrons and VLDL are another source of preβ-HDL. Hepatic lipase activity is increased by androgens and decreased by estrogens, which may account for higher concentrations of plasma HDL2 in women.
HDL concentrations vary reciprocally with plasma triacylglycerol concentrations and directly with the activity of lipoprotein lipase. This may be due to surplus surface constituents, eg, phospholipid and apo A-I, being released during hydrolysis of chylomicrons and VLDL and contributing toward the formation of preβ-HDL and discoidal HDL. HDL2 concentrations are inversely related to the incidence of atherosclerosis, possibly because they reflect the efficiency of reverse cholesterol transport. HDLc (HDL1) is found in the blood of diet-induced hypercholesterolemic animals. It is rich in cholesterol, and its sole apolipoprotein is apo E. It appears that all plasma lipoproteins are interrelated components of one or more metabolic cycles that together are responsible for the complex process of plasma lipid transport.
THE LIVER PLAYS A CENTRAL ROLE IN LIPID TRANSPORT & METABOLISM
The liver carries out the following major functions in lipid metabolism:
1. It facilitates the digestion and absorption of lipids by the production of bile, which contains cholesterol and bile salts synthesized within the liver de novo or after uptake of lipoprotein cholesterol (Chapter 26).
2. It actively synthesizes and oxidizes fatty acids (Chapters 22 & 23) and also synthesizes triacylglycerols and phospholipids (Chapter 24).
3. It converts fatty acids to ketone bodies (ketogenesis) (Chapter 22).
4. It plays an integral part in the synthesis and metabolism of plasma lipoproteins (this chapter).
Hepatic VLDL Secretion Is Related to Dietary & Hormonal Status
The cellular events involved in VLDL formation and secretion have been described above (Figure 25–2) and are shown in Figure 25–6. Hepatic triacylglycerol synthesis provides the immediate stimulus for the formation and secretion of VLDL. The fatty acids used are derived from two possible sources: (1) synthesis within the liver from acetyl-CoA derived mainly from carbohydrate (perhaps not so important in humans) and (2) uptake of FFA from the circulation. The first source is predominant in the well-fed condition, when fatty acid synthesis is high and the level of circulating FFA is low. As triacylglycerol does not normally accumulate in the liver in these conditions, it must be inferred that it is transported from the liver in VLDL as rapidly as it is synthesized. FFA from the circulation are the main source during starvation, the feeding of high-fat diets, or in diabetes mellitus, when hepatic lipogenesis is inhibited. Synthesis of VLDL takes place in the endoplasmic reticulum (ER) and requires the microsomal triacylglycerol transfer proten (MTP), which transfers triacylglycerol from the cytosol into the ER lumen where it is incorporated into particles with cholesterol, phospholipids and apoB-100 (Figure 25–6). Factors that enhance both the synthesis of triacylglycerol and the secretion of VLDL by the liver include (1) the fed state rather than the starved state; (2) the feeding of diets high in carbohydrate (particularly if they contain sucrose or fructose), leading to high rates of lipogenesis and esterification of fatty acids; (3) high levels of circulating FFA; (4) ingestion of ethanol; and (5) the presence of high concentrations of insulin and low concentrations of glucagon, which enhance fatty acid synthesis and esterification and inhibit their oxidation (Figure 25–6).
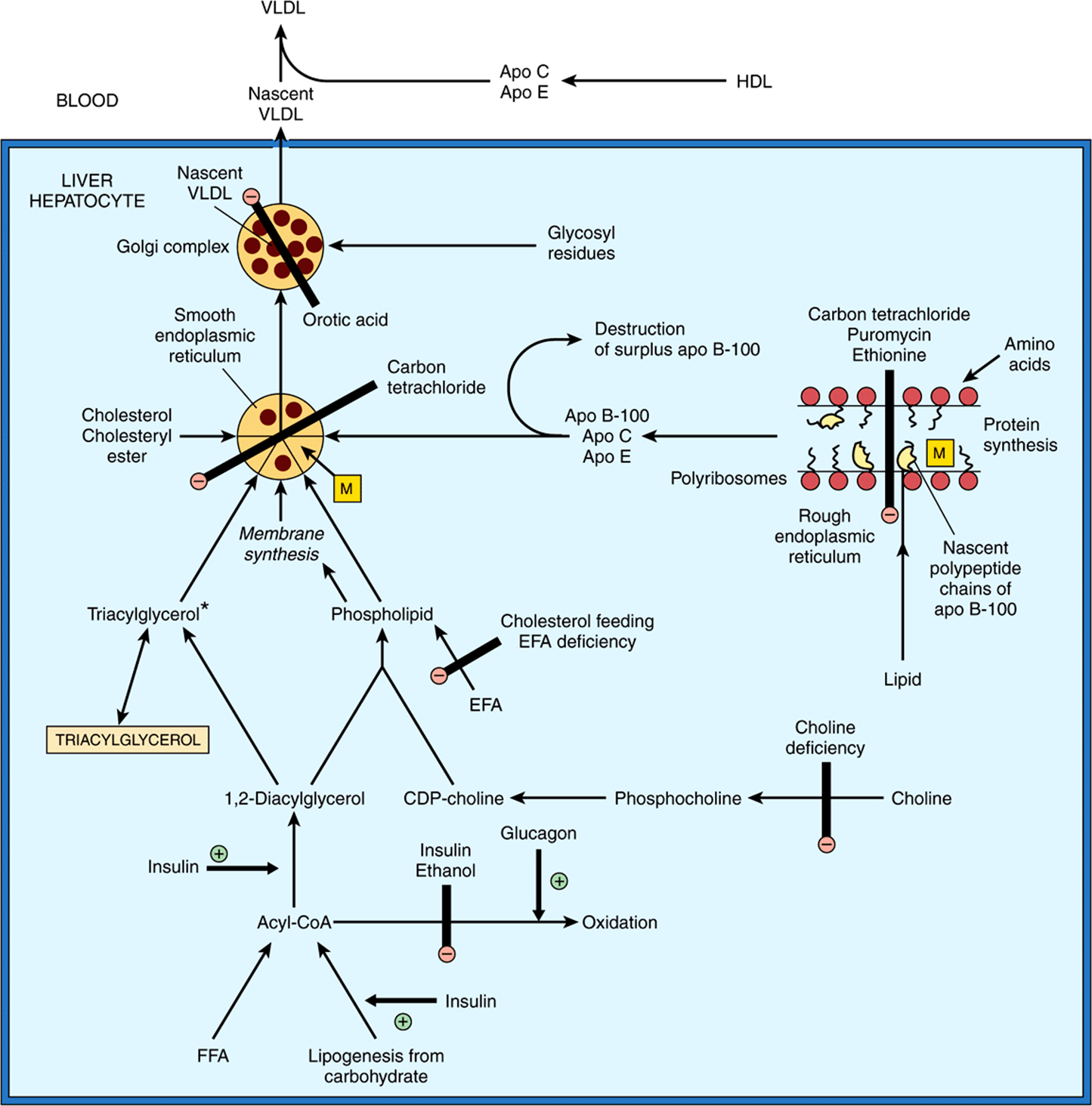
FIGURE 25–6 The synthesis of very low density lipoprotein (VLDL) in the liver and the possible loci of action of factors causing accumulation of triacylglycerol and fatty liver. (Apo, apolipoprotein; EFA, essential fatty acids; FFA, free fatty acids; HDL, high-density lipoproteins; M, microsomal triacylglycerol transfer protein.) The pathways indicated form a basis for events depicted in Figure 25–2. The main triacylglycerol pool in liver is not on the direct pathway of VLDL synthesis from acyl-CoA. Thus, FFA, insulin, and glucagon have immediate effects on VLDL secretion as their effects impinge directly on the small triacylglycerol precursor pool*. In the fully fed state, apo B-100 is synthesized in excess of requirements for VLDL secretion and the surplus is destroyed in the liver. During translation of apo B-100 in the rough endoplasmic reticulum, microsomal transfer protein-mediated lipid transport enables lipid to become associated with the nascent polypeptide chain. After release from the ribosomes, these particles fuse with more lipids from the smooth endoplasmic reticulum, producing nascent VLDL.
CLINICAL ASPECTS
Imbalance in the Rate of Triacylglycerol Formation & Export Causes Fatty Liver
For a variety of reasons, lipid—mainly as triacylglycerol—can accumulate in the liver (Figure 25–6). Extensive accumulation is regarded as a pathologic condition. Nonalcoholic fatty liver disease (NAFLD) is the most common liver disorder worldwide. When accumulation of lipid in the liver becomes chronic, inflammatory and fibrotic changes may develop leading to nonalcoholic steatohepatitis (NASH), which can progress to liver diseases including cirrhosis, hepatocarcinoma, and liver failure.
Fatty livers fall into two main categories. The first type is associated with raised levels of plasma free fatty acids resulting from mobilization of fat from adipose tissue or from the hydrolysis of lipoprotein triacylglycerol by lipoprotein lipase in extrahepatic tissues. The production of VLDL does not keep pace with the increasing influx and esterification of free fatty acids, allowing triacylglycerol to accumulate, which in turn causes a fatty liver. This occurs during starvation and the feeding of high-fat diets. The ability to secrete VLDL may also be impaired (eg, in starvation). In uncontrolled diabetes mellitus, twin lamb disease, and ketosis in cattle, fatty infiltration is sufficiently severe to cause visible pallor (fatty appearance) and enlargement of the liver with possible liver dysfunction.
The second type of fatty liver is usually due to a metabolic block in the production of plasma lipoproteins, thus allowing triacylglycerol to accumulate. Theoretically, the lesion may be due to (1) a block in apolipoprotein synthesis (or an increase in its degradation before it can be incorporated into VLDL), (2) a block in the synthesis of the lipoprotein from lipid and apolipoprotein, (3) a failure in provision of phospholipids that are found in lipoproteins, or (4) a failure in the secretory mechanism itself.
One type of fatty liver that has been studied extensively in rats is caused by a deficiency of choline, which has therefore been called a lipotropic factor. The antibiotic puromycin, ethionine (α-amino-γ-mercaptobutyric acid), carbon tetrachloride, chloroform, phosphorus, lead, and arsenic all cause fatty liver and a marked reduction in concentration of VLDL in rat blood. Choline will not protect the organism against these agents, but appears to aid in recovery. The action of carbon tetrachloride probably involves formation of free radicals causing lipid peroxidation. Some protection against this is provided by the antioxidant action of vitamin E-supplemented diets. The action of ethionine is thought to be caused by a reduction in availability of ATP due to its replacing methionine in S-adenosylmethionine, trapping available adenine and preventing synthesis of ATP. Orotic acid also causes fatty liver; it is believed to interfere with glycosylation of the lipoprotein, thus inhibiting release, and may also impair the recruitment of triacylglycerol to the particles. A deficiency of vitamin E enhances the hepatic necrosis of the choline deficiency type of fatty liver. Added vitamin E or a source of selenium has a protective effect by combating lipid peroxidation. In addition to protein deficiency, essential fatty acid and vitamin deficiencies (eg, linoleic acid, pyridoxine, and pantothenic acid) can cause fatty infiltration of the liver.
Ethanol Also Causes Fatty Liver
Alcoholic fatty liver is the first stage in alcoholic liver disease (ALD) which is caused by alcoholism and ultimately leads to cirrhosis. The fat accumulation in the liver is caused by a combination of impaired fatty acid oxidation and increased lipogenesis, which is thought to be due to changes in the [NADH]/[NAD+] redox potential in the liver, and also to interference with the action of transcription factors regulating the expression of the enzymes involved in the pathways. Oxidation of ethanol by alcohol dehydrogenase leads to excess production of NADH, which competes with reducing equivalents from other substrates, including fatty acids, for the respiratory chain. This inhibits their oxidation and causes increased esterification of fatty acids to form triacylglycerol, resulting in the fatty liver. Oxidation of ethanol leads to the formation of acetaldehyde, which is oxidized by aldehyde dehydrogenase, producing acetate. The increased (NADH)/(NAD+) ratio also causes increased (lactate)/(pyruvate), resulting in hyperlacticacidemia, which decreases excretion of uric acid, aggravating gout.
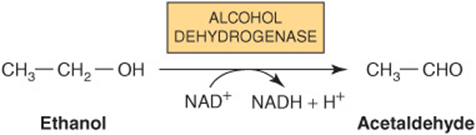
Some metabolism of ethanol takes place via a cytochrome P450-dependent microsomal ethanol oxidizing system (MEOS) involving NADPH and O2. This system increases in activity in chronic alcoholism and may account for the increased metabolic clearance in this condition. Ethanol also inhibits the metabolism of some drugs, eg, barbiturates, by competing for cytochrome P450-dependent enzymes.
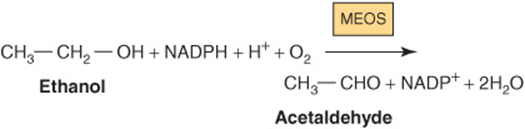
In some Asian populations and Native Americans, alcohol consumption results in increased adverse reactions to acetaldehyde owing to a genetic defect of mitochondrial aldehyde dehydrogenase.
ADIPOSE TISSUE IS THE MAIN STORE OF TRIACYLGLYCEROL IN THE BODY
Triacylglycerols are stored in adipose tissue in large lipid droplets and are continually undergoing lipolysis (hydrolysis) and re-esterification (Figure 25–7). These two processes are entirely different pathways involving different reactants and enzymes. This allows the processes of esterification or lipolysis to be regulated separately by many nutritional, metabolic, and hormonal factors. The balance between these two processes determines the magnitude of the FFA pool in adipose tissue, which in turn determines the level of FFA circulating in the plasma. Since the latter has most profound effects upon the metabolism of other tissues, particularly liver and muscle, the factors operating in adipose tissue that regulate the outflow of FFA exert an influence far beyond the tissue itself.
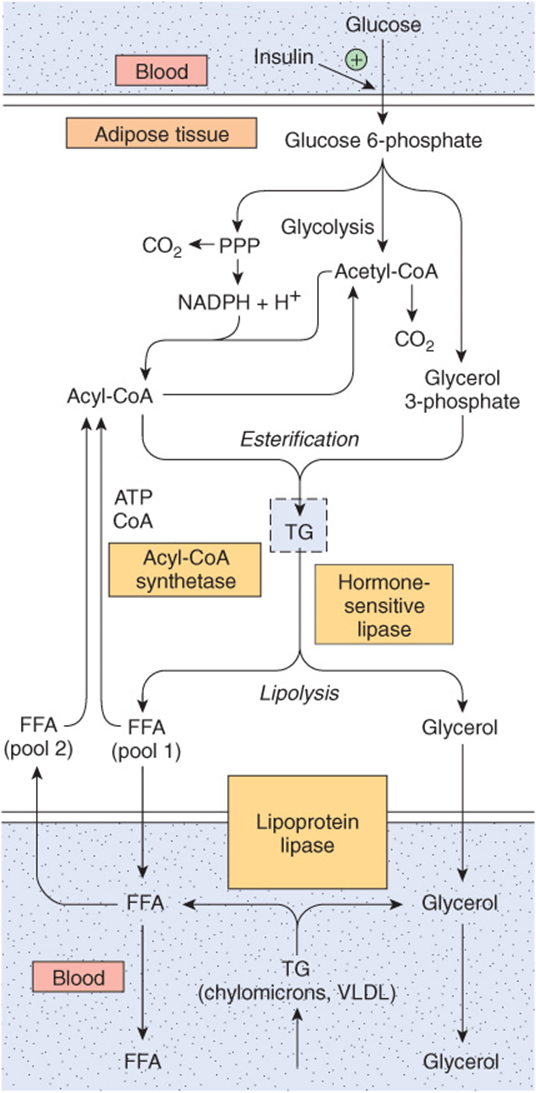
FIGURE 25–7 Triacylglycerol metabolism in adipose tissue. Hormone-sensitive lipase is activated by ACTH, TSH, glucagon, epinephrine, norepinephrine, and vasopressin and inhibited by insulin, prostaglandin E1, and nicotinic acid. Details of the formation of glycerol 3-phosphate from intermediates of glycolysis are shown in Figure 24–2. (FFA, free fatty acids; PPP, pentose phosphate pathway; TG, triacylglycerol; VLDL, very low density lipoprotein.)
The Provision of Glycerol 3-Phosphate Regulates Esterification: Lipolysis Is Controlled by Hormone-Sensitive Lipase
Triacylglycerol is synthesized from acyl-CoA and glycerol 3-phosphate (Figure 24–2). Since the enzyme glycerol kinase is not expressed in adipose tissue, glycerol cannot be utilized for the provision of glycerol 3-phosphate, which must be supplied from glucose via glycolysis.
Triacylglycerol undergoes hydrolysis by a hormone-sensitive lipase to form FFA and glycerol. This lipase is distinct from lipoprotein lipase, which catalyzes lipoprotein triacylglycerol hydrolysis before its uptake into extrahepatic tissues (see above). Since the glycerol cannot be utilized, it enters the blood and is taken up and transported to tissues such as the liver and kidney, which possess an active glycerol kinase. The FFA formed by lipolysis can be reconverted in adipose tissue to acyl-CoA by acyl-CoA synthetase and reesterified with glycerol 3-phosphate to form triacylglycerol. Thus, there is a continuous cycle of lipolysis and re-esterification within the tissue.However, when the rate of re-esterification is not sufficient to match the rate of lipolysis, FFA accumulate and diffuse into the plasma, where they bind to albumin and raise the concentration of plasma-free fatty acids.
Increased Glucose Metabolism Reduces the Output of FFA
When the utilization of glucose by adipose tissue is increased, the FFA outflow decreases. However, the release of glycerol continues, demonstrating that the effect of glucose is not mediated by reducing the rate of lipolysis. The effect is due to the provision of glycerol 3-phosphate, which enhances esterification of FFA. Glucose can take several pathways in adipose tissue, including oxidation to CO2 via the citric acid cycle, oxidation in the pentose phosphate pathway, conversion to long-chain fatty acids, and formation of acylglycerol via glycerol 3-phosphate (Figure 25–7). When glucose utilization is high, a larger proportion of the uptake is oxidized to CO2 and converted to fatty acids. However, as total glucose utilization decreases, the greater proportion of the glucose is directed to the formation of glycerol 3-phosphate for the esterification of acylCoA, which helps to minimize the efflux of FFA.
HORMONES REGULATE FAT MOBILIZATION
Adipose Tissue Lipolysis Is Inhibited by Insulin
The rate of release of FFA from adipose tissue is affected by many hormones that influence either the rate of esterification or the rate of lipolysis. Insulin inhibits the release of FFA from adipose tissue, which is followed by a fall in circulating plasma free fatty acids. Insulin also enhances lipogenesis and the synthesis of acylglycerol and increases the oxidation of glucose to CO2 via the pentose phosphate pathway. All of these effects are dependent on the presence of glucose and can be explained, to a large extent, on the basis of the ability of insulin to enhance the uptake of glucose into adipose cells via the GLUT 4 transporter. In addition, insulin increases the activity of the enzymes pyruvate dehydrogenase, acetyl-CoA carboxylase, and glycerol phosphate acyltransferase, reinforcing the effects of increased glucose uptake on the enhancement of fatty acid and acylglycerol synthesis. These three enzymes are regulated in a coordinate manner by phosphorylation-dephosphorylation mechanisms.
Another principal action of insulin in adipose tissue is to inhibit the activity of hormone-sensitive lipase, reducing the release not only of FFA but also of glycerol. Adipose tissue is much more sensitive to insulin than many other tissues, which points to adipose tissue as a major site of insulin action in vivo.
Several Hormones Promote Lipolysis
Other hormones accelerate the release of FFA from adipose tissue and raise the plasma-free fatty acid concentration by increasing the rate of lipolysis of the triacylglycerol stores (Figure 25–8). These include epinephrine, norepinephrine, glucagon, adrenocorticotropic hormone (ACTH), α- and β-melanocyte-stimulating hormones (MSH), thyroid-stimulating hormone (TSH), growth hormone (GH), and vasopressin. Many of these activate hormone-sensitive lipase. For an optimal effect, most of these lipolytic processes require the presence of glucocorticoids and thyroid hormones. These hormones act in a facilitatory or permissive capacity with respect to other lipolytic endocrine factors.
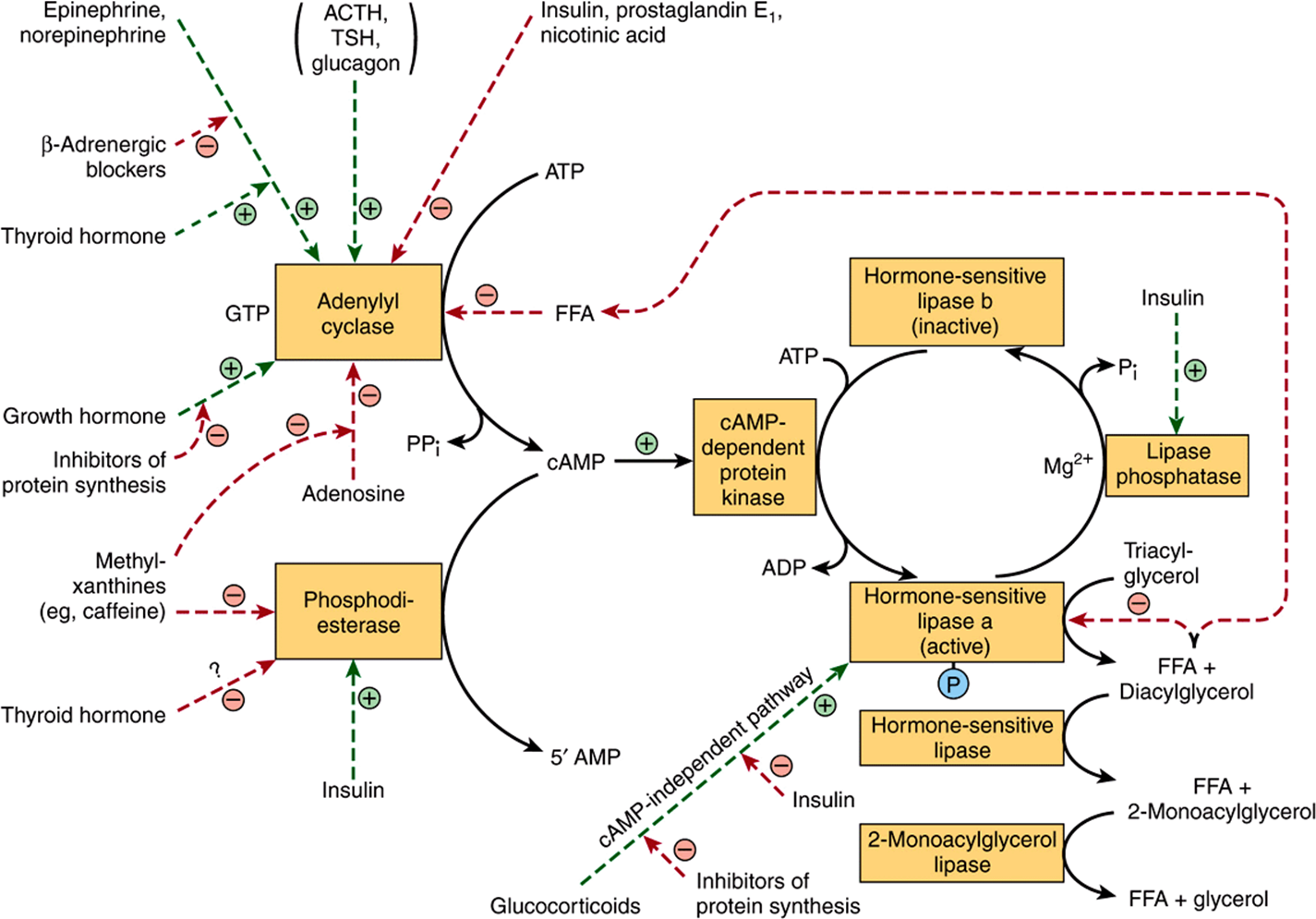
FIGURE 25–8 Control of adipose tissue lipolysis. (FFA, free fatty acids; TSH, thyroid-stimulating hormone.) Note the cascade sequence of reactions affording amplification at each step. The lipolytic stimulus is “switched off” by removal of the stimulating hormone; the action of lipase phosphatase; the inhibition of the lipase and adenylyl cyclase by high concentrations of FFA; the inhibition of adenylyl cyclase by adenosine; and the removal of cAMP by the action of phosphodiesterase. ACTH, TSH, and glucagon may not activate adenylyl cyclase in vivo since the concentration of each hormone required in vitro is much higher than is found in the circulation. Positive ![]() and negative
and negative ![]() regulatory effects are represented by broken lines and substrate flow by solid lines.
regulatory effects are represented by broken lines and substrate flow by solid lines.
The hormones that act rapidly in promoting lipolysis, ie, catecholamines (epinephrine and nor-epinephrine), do so by stimulating the activity of adenylyl cyclase, the enzyme that converts ATP to cAMP. The mechanism is analogous to that responsible for hormonal stimulation of glycogenolysis (Chapter 19). cAMP, by stimulating cAMP-dependent protein kinase, activates hormone-sensitive lipase. Thus, processes which destroy or preserve cAMP influence lipolysis. cAMP is degraded to 5′-AMP by the enzyme cyclic 3’,5’-nucleotide phosphodiesterase. This enzyme is inhibited by methylxanthines such as caffeine and theophylline. Insulin antagonizes the effect of the lipolytic hormones. Lipolysis appears to be more sensitive to changes in concentration of insulin than are glucose utilization and esterification. The antilipolytic effects of insulin, nicotinic acid, and prostaglandin E1 are accounted for by inhibition of the synthesis of cAMP at the adenylyl cyclase site, acting through a Gi protein. Insulin also stimulates phosphodiesterase and the lipase phosphatase that inactivates hormone-sensitive lipase. The effect of growth hormone in promoting lipolysis is dependent on synthesis of proteins involved in the formation of cAMP. Glucocorticoids promote lipolysis via synthesis of new lipase protein by a cAMP-independent pathway, which may be inhibited by insulin, and also by promoting transcription of genes involved in the cAMP signal cascade. These findings help to explain the role of the pituitary gland and the adrenal cortex in enhancing fat mobilization. Adipose tissue secretes hormones such as adiponectin, which modulates glucose and lipid metabolism in muscle and liver, and leptin, which regulates energy homeostasis. Current evidence suggests that the main role of leptin in humans is to suppress appetite when food intake is sufficient. If it is lacking, food intake may be uncontrolled, causing obesity.
The sympathetic nervous system, through liberation of norepinephrine in adipose tissue, plays a central role in the mobilization of FFA. Thus, the increased lipolysis caused by many of the factors described above can be reduced or abolished by denervation of adipose tissue or by ganglionic blockade.
Perilipin Regulates the Balance Between Triacylglycerol Storage and Lipolysis in Adipocytes
Perilipin, a protein involved in the formation of lipid droplets in adipocytes, inhibits lipolysis in basal conditions by preventing access of the lipase enzymes to the stored triacylglycerols. On stimulation with hormones which promote triacylglycerol degradation, however, the protein targets hormone-sensitive lipase to the lipid droplet surface and thus promotes lipolysis. Perilipin, therefore, enables the storage and breakdown of triacylglycerol to be coordinated according to the metabolic needs of the body.
Human Adipose Tissue May Not Be an Important Site of Lipogenesis
In adipose tissue, there is no significant incorporation of glucose or pyruvate into long-chain fatty acids, ATP-citrate lyase, a key enzyme in lipogenesis, does not appear to be present, and other lipogenic enzymes—eg, glucose-6-phosphate dehydrogenase and the malic enzyme—do not undergo adaptive changes. Indeed, it has been suggested that in humans there is a “carbohydrate excess syndrome” due to a unique limitation in ability to dispose of excess carbohydrate by lipogenesis. In birds, lipogenesis is confined to the liver, where it is particularly important in providing lipids for egg formation, stimulated by estrogens.
BROWN ADIPOSE TISSUE PROMOTES THERMOGENESIS
Brown adipose tissue is involved in metabolism, particularly at times when heat generation is necessary. Thus, the tissue is extremely active in some species, for example, during arousal from hibernation, in animals exposed to cold (nonshivering thermogenesis), and in heat production in the newborn. Though not a prominent tissue in humans, it is present in normal individuals, where it could be responsible for “diet-induced thermogenesis.” It is noteworthy that brown adipose tissue is reduced or absent in obese persons. The tissue is characterized by a well-developed blood supply and a high content of mitochondria and cytochromes, but low activity of ATP synthase. Metabolic emphasis is placed on oxidation of both glucose and fatty acids. Norepinephrine liberated from sympathetic nerve endings is important in increasing lipolysis in the tissue and increasing synthesis of lipoprotein lipase to enhance utilization of triacylglycerol-rich lipoproteins from the circulation. Oxidation and phosphorylation are not coupled in mitochondria of this tissue, and the phosphorylation that does occur is at the substrate level, eg, at the succinate thiokinase step and in glycolysis. Thus, oxidation produces much heat, and little free energy is trapped in ATP. A thermogenic uncoupling protein, thermogenin, acts as a proton conductance pathway dissipating the electrochemical potential across the mitochondrial membrane (Figure 25–9).
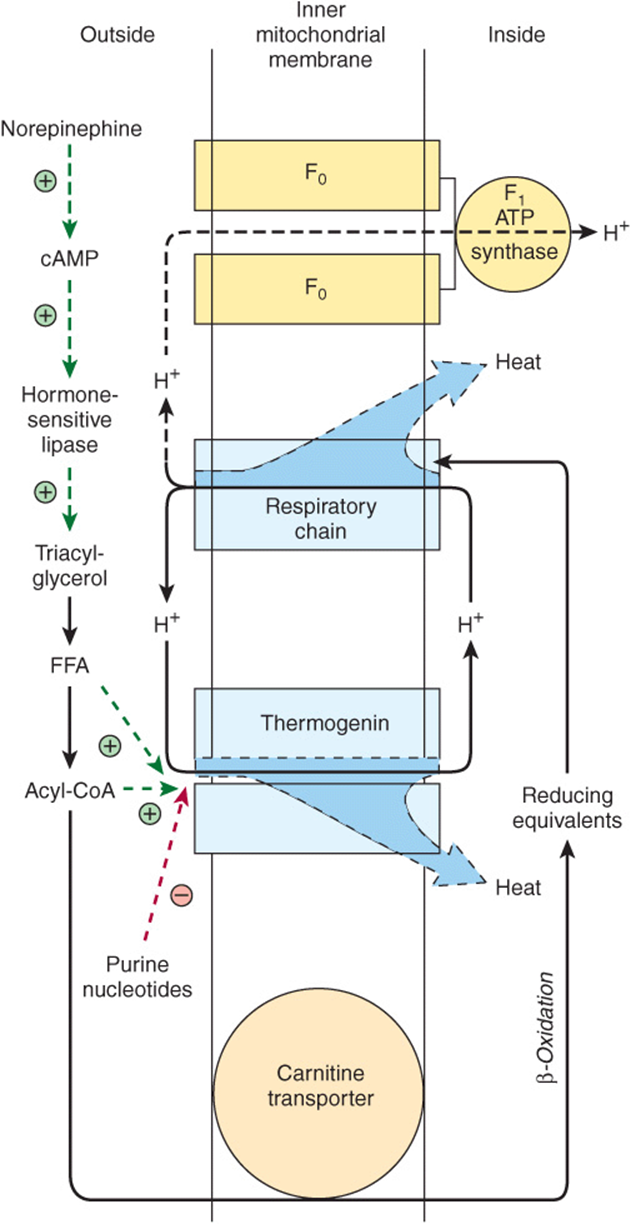
FIGURE 25–9 Thermogenesis in brown adipose tissue. Activity of the respiratory chain produces heat in addition to translocating protons (Chapter 13). These protons dissipate more heat when returned to the inner mitochondrial compartment via thermogenin instead of via the F1 ATP synthase, the route that generates ATP (Figure 13–7). The passage of H+ via thermogenin is inhibited by purine nucleotides when brown adipose tissue is unstimulated. Under the influence of norepinephrine, the inhibition is removed by the production of free fatty acids (FFA) and acyl-CoA. Note the dual role of acyl-CoA in both facilitating the action of thermogenin and supplying reducing equivalents for the respiratory chain. ![]() and
and ![]() signify positive or negative regulatory effects.
signify positive or negative regulatory effects.
SUMMARY
![]() Since nonpolar lipids are insoluble in water, for transport between the tissues in the aqueous blood plasma they are combined with amphipathic lipids and proteins to make water-miscible lipoproteins.
Since nonpolar lipids are insoluble in water, for transport between the tissues in the aqueous blood plasma they are combined with amphipathic lipids and proteins to make water-miscible lipoproteins.
![]() Four major groups of lipoproteins are recognized. Chylomicrons transport lipids resulting from digestion and absorption. Very low density lipoproteins (VLDL) transport triacylglycerol from the liver. Low-density lipoproteins (LDL) deliver cholesterol to the tissues, and high-density lipoproteins (HDL) remove cholesterol from the tissues and return it to the liver for excretion in the process known as reverse cholesterol transport.
Four major groups of lipoproteins are recognized. Chylomicrons transport lipids resulting from digestion and absorption. Very low density lipoproteins (VLDL) transport triacylglycerol from the liver. Low-density lipoproteins (LDL) deliver cholesterol to the tissues, and high-density lipoproteins (HDL) remove cholesterol from the tissues and return it to the liver for excretion in the process known as reverse cholesterol transport.
![]() Chylomicrons and VLDL are metabolized by hydrolysis of their triacylglycerol, and lipoprotein remnants are left in the circulation. These are taken up by liver, but some of the remnants (IDL), resulting from VLDL form LDL, which is taken up by the liver and other tissues via the LDL receptor.
Chylomicrons and VLDL are metabolized by hydrolysis of their triacylglycerol, and lipoprotein remnants are left in the circulation. These are taken up by liver, but some of the remnants (IDL), resulting from VLDL form LDL, which is taken up by the liver and other tissues via the LDL receptor.
![]() Apolipoproteins constitute the protein moiety of lipoproteins. They act as enzyme activators (eg, apo C-II and apo A-I) or as ligands for cell receptors (eg, apo A-I, apo E, and apo B-100).
Apolipoproteins constitute the protein moiety of lipoproteins. They act as enzyme activators (eg, apo C-II and apo A-I) or as ligands for cell receptors (eg, apo A-I, apo E, and apo B-100).
![]() Triacylglycerol is the main storage lipid in adipose tissue. Upon mobilization, FFA and glycerol are released. FFA are an important fuel source.
Triacylglycerol is the main storage lipid in adipose tissue. Upon mobilization, FFA and glycerol are released. FFA are an important fuel source.
![]() Brown adipose tissue is the site of “nonshivering thermogenesis.” It is found in hibernating and newborn animals and is present in small quantity in humans. Thermogenesis results from the presence of an uncoupling protein, thermogenin, in the inner mitochondrial membrane.
Brown adipose tissue is the site of “nonshivering thermogenesis.” It is found in hibernating and newborn animals and is present in small quantity in humans. Thermogenesis results from the presence of an uncoupling protein, thermogenin, in the inner mitochondrial membrane.
REFERENCES
Arner P: Human fat cell lipolysis: biochemistry, regulation and clinical role. Best Pract Res Clin Endocrinol Metab 2005;19:471.
Brasaemle DL: Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J Lipid Res 2007;48:2547.
Fielding CJ, Fielding PE: Dynamics of lipoprotein transport in the circulatory system. In Biochemistry of Lipids, Lipoproteins and Membranes, 5th ed. Vance DE, Vance JE (editors). Elsevier, 2008;533-554.
Goldberg IJ, Merkel M: Lipoprotein lipase: physiology, biochemistry and molecular biology. Front Biosci 2001;6:D388.
Kershaw EE, Flier JS: Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 2004;89:2548.
Lass A, Zimmermann R, Oberer M, et al: Lipolysis—a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog Lipid Res 2011;50:14.
Lenz A, Diamond FB: Obesity: the hormonal milieu. Curr Opin Endocrinol Diabetes Obes 2008;15:9. Redgrave TG: Chylomicron metabolism. Biochem Soc Trans 2004;32:79.
Schreuder TC, Verwer BJ, van Nieuwkerk CM, et al: Nonalcoholic fatty liver disease: an overview of current insights in pathogenesis, diagnosis and treatment. World J Gastroenterol 2008;14:2474.
Sell H, Deshaies Y, Richard D: The brown adipocyte: update on its metabolic role. Int J Biochem Cell Biol 2004;36:2098.
Vance JE, Adeli K: Assembly and secretion of triacylglycerol-rich lipoproteins. In Biochemistry of Lipids, Lipoproteins and Membranes, 5th ed. Vance DE, Vance JE (editors). Elsevier, 2008;507-532.