Harper’s Illustrated Biochemistry, 29th Edition (2012)
SECTION III. Metabolism of Proteins & Amino Acids
Chapter 30. Conversion of Amino Acids to Specialized Products
Victor W. Rodwell, PhD
OBJECTIVES
After studying this chapter, you should be able to:
![]() Describe how amino acids participate in a variety of biosynthetic processes other than protein synthesis.
Describe how amino acids participate in a variety of biosynthetic processes other than protein synthesis.
![]() Outline how arginine participates in the biosynthesis of creatine, nitric oxide (NO), putrescine, spermine, and spermidine.
Outline how arginine participates in the biosynthesis of creatine, nitric oxide (NO), putrescine, spermine, and spermidine.
![]() Indicate the contribution of cysteine and of β-alanine to the structure of coenzyme A and of cysteine to the structure of taurocholic acid.
Indicate the contribution of cysteine and of β-alanine to the structure of coenzyme A and of cysteine to the structure of taurocholic acid.
![]() Discuss the role of glycine in drug catabolism.
Discuss the role of glycine in drug catabolism.
![]() Document the role of glycine in the biosynthesis of heme, purines, creatine, and sarcosine.
Document the role of glycine in the biosynthesis of heme, purines, creatine, and sarcosine.
![]() Identify the reaction that converts an amino acid to the neurotransmitter histamine.
Identify the reaction that converts an amino acid to the neurotransmitter histamine.
![]() Document the role of S-adenosylmethionine as a source of methyl groups in metabolism.
Document the role of S-adenosylmethionine as a source of methyl groups in metabolism.
![]() Recognize the tryptophan metabolites serotonin and melatonin, and the conversion of tryptophan to tryptamine and subsequently to indole 3-acetate.
Recognize the tryptophan metabolites serotonin and melatonin, and the conversion of tryptophan to tryptamine and subsequently to indole 3-acetate.
![]() Indicate the role of tyrosine in the formation of norepinephrine, epinephrine, triiodothyronine, and thyroxine.
Indicate the role of tyrosine in the formation of norepinephrine, epinephrine, triiodothyronine, and thyroxine.
![]() Illustrate the key roles of peptidyl serine, threonine and tyrosine in metabolic regulation and signal transduction pathways.
Illustrate the key roles of peptidyl serine, threonine and tyrosine in metabolic regulation and signal transduction pathways.
![]() Outline the roles of glycine, arginine, and S-adenosylmethionine in the biosynthesis of creatine.
Outline the roles of glycine, arginine, and S-adenosylmethionine in the biosynthesis of creatine.
![]() Describe the role of creatine phosphate in energy homeostasis.
Describe the role of creatine phosphate in energy homeostasis.
![]() Describe the formation of γ-aminobutyrate (GABA) and the rare metabolic disorders associated with defects in GABA catabolism.
Describe the formation of γ-aminobutyrate (GABA) and the rare metabolic disorders associated with defects in GABA catabolism.
BIOMEDICAL IMPORTANCE
Certain proteins contain amino acids that have been post-translationally modified to permit them to perform specific functions. Examples include the carboxylation of glutamate to form γ-carboxyglutamate, which functions in Ca2+binding, the hydroxylation of proline for incorporation into the collagen triple helix, and the hydroxylation of lysine to hydroxylysine, whose subsequent modification and cross-linking stabilizes maturing collagen fibers. In addition to serving as the building blocks for protein synthesis, amino acids serve as precursors of biologic materials such as heme, purines, pyrimidines, hormones, neurotransmitters, and biologically active peptides. Histamine plays a central role in many allergic reactions. Neurotransmitters derived from amino acids include γ-aminobutyrate, 5-hydroxytryptamine (serotonin), dopamine, norepinephrine, and epinephrine. Many drugs used to treat neurologic and psychiatric conditions act by altering the metabolism of these neurotransmitters. Discussed below are the metabolism and metabolic roles of selected α- and non-α amino acids.
L-α-AMINO ACIDS
Alanine
Alanine serves as a carrier of ammonia and of the carbons of pyruvate from skeletal muscle to liver via the Cori cycle (see Figure 20–4), and together with glycine constitutes a major fraction of the free amino acids in plasma.
Arginine
Figure 30–1 summarizes the metabolic fates of arginine. In addition to serving as a carrier of nitrogen atoms in urea biosynthesis (see Figure 28–13), the guanidino group of arginine is incorporated into creatine, and following conversion to ornithine, its carbon skeleton becomes that of the polyamines putrescine and spermine.
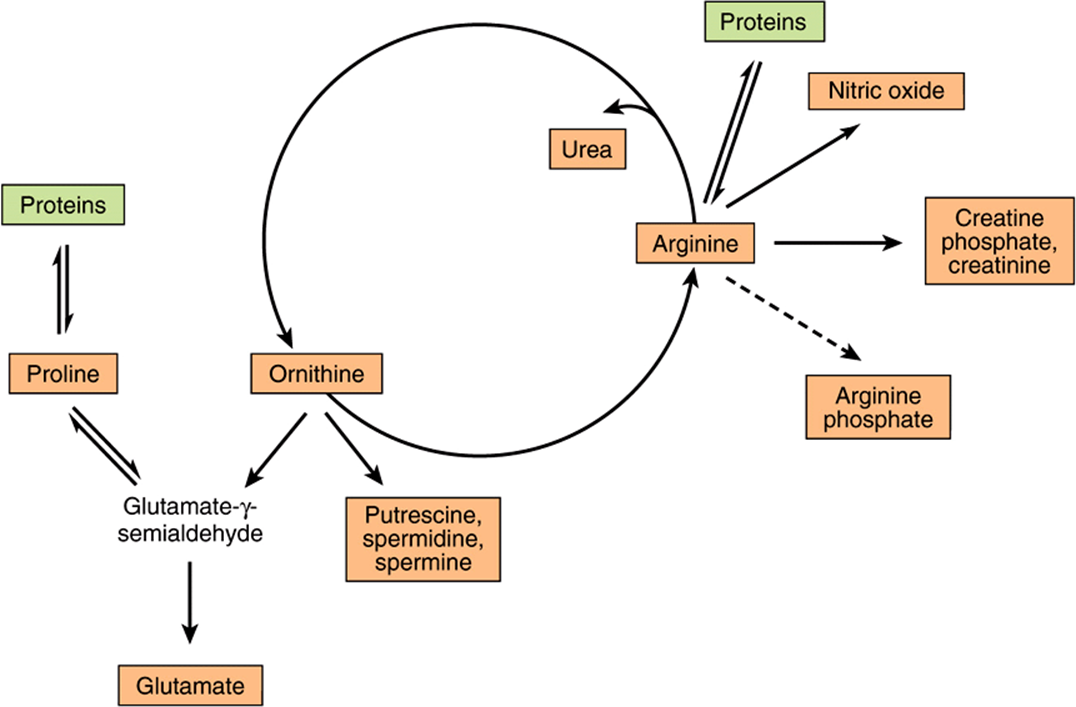
FIGURE 30–1 Arginine, ornithine, and proline metabolism. Reactions with solid arrows all occur in mammalian tissues. Putrescine and spermine synthesis occurs in both mammals and bacteria. Arginine phosphate of invertebrate muscle functions as a phosphagen analogous to creatine phosphate of mammalian muscle.
The reaction catalyzed by NO synthase (Figure 30–2), a five-electron oxidoreductase with multiple cofactors, converts one nitrogen of the guanidine group of arginine to citrulline and NO, an intercellular signaling molecule that serves as a neurotransmitter, smooth muscle relaxant, and vasodilator (see Chapter 49).
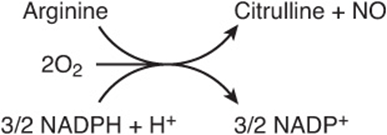
FIGURE 30–2 The reaction catalyzed by nitric oxide synthase.
Cysteine
Cysteine participates in the biosynthesis of coenzyme A (see Figure 44–18) by reacting with pantothenate to form 4-phosphopantothenoyl-cysteine (Figure 30–3). Three enzyme-catalyzed reactions convert cysteine to taurine, which can displace the coenzyme A moiety of cholyl-CoA to form the bile acid taurocholic acid (see Figure 26–7). The conversion of cysteine to taurine is initiated by its oxidation to cysteine sulfinate, catalyzed by the nonheme Fe2+enzyme cysteine dioxygenase. Decarboxylation of cysteine sulfinate by cysteine sulfinate decarboxylase forms hypotaurine, whose oxidation by hypotaurine dehydrogenase forms taurine (Figure 30–4).
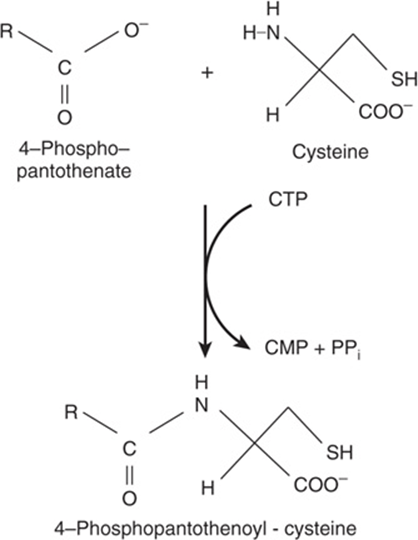
FIGURE 30–3 The reaction catalyzed by phosphopantothenate cysteine ligase.
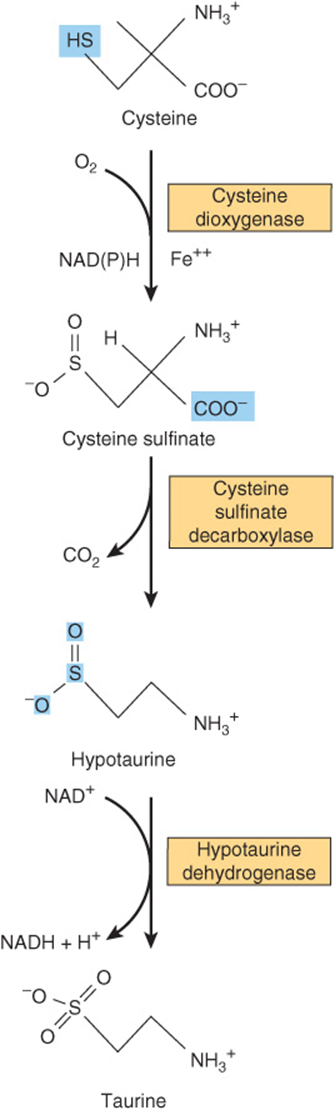
FIGURE 30–4 Conversion of cysteine to taurine. The reactions are catalyzed by cysteine dioxygenase, cysteine sulfinate decarboxylase, and hypotaurine decarboxylase, respectively.
Glycine
Metabolites and pharmaceuticals excreted as water-soluble glycine conjugates include glycocholic acid (see Chapter 26) and hippuric acid formed from the food additive benzoate (Figure 30–5). Many drugs, drug metabolites, and other compounds with carboxyl groups are excreted in the urine as glycine conjugates. Glycine is incorporated into creatine, and the nitrogen and α-carbon of glycine are incorporated into the pyrrole rings and the methylene bridge carbons of heme (see Chapter 31), and the entire glycine molecule becomes atoms 4, 5, and 7 of purines (see Figure 33–1).
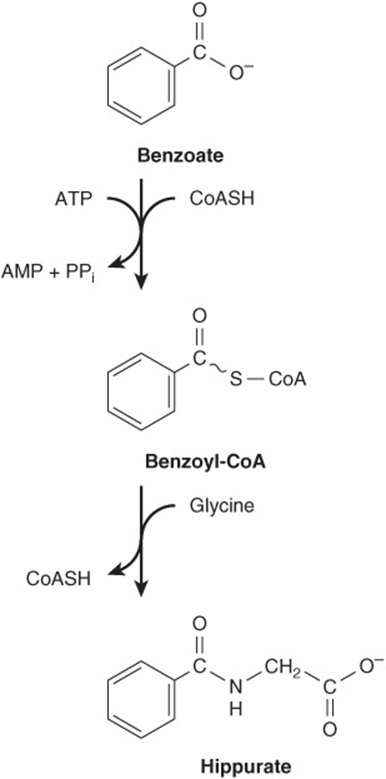
FIGURE 30–5 Biosynthesis of hippurate. Analogous reactions occur with many acidic drugs and catabolites.
Histidine
Decarboxylation of histidine by the pyridoxal 5′-phosphate-dependent enzyme histidine decarboxylase forms histamine (Figure 30–6). A biogenic amine that functions in allergic reactions and gastric secretion, histamine is present in all tissues. Its concentration in the brain hypothalamus varies in accordance with a circadian rhythm. Histidine compounds present in the human body include ergothioneine, carnosine, and dietary anserine (Figure 30–7). While their physiological functions are unknown, carnosine (β-alanyl-histidine) and homocarnosine (γ-aminobutyryl-histidine) are major constituents of excitable tissues, brain, and skeletal muscle. Urinary levels of 3-methylhistidine are unusually low in patients with Wilson’s disease.
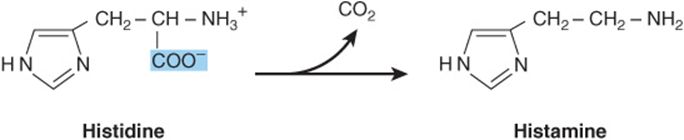
FIGURE 30–6 The reaction catalyzed by histidine decarboxylase.
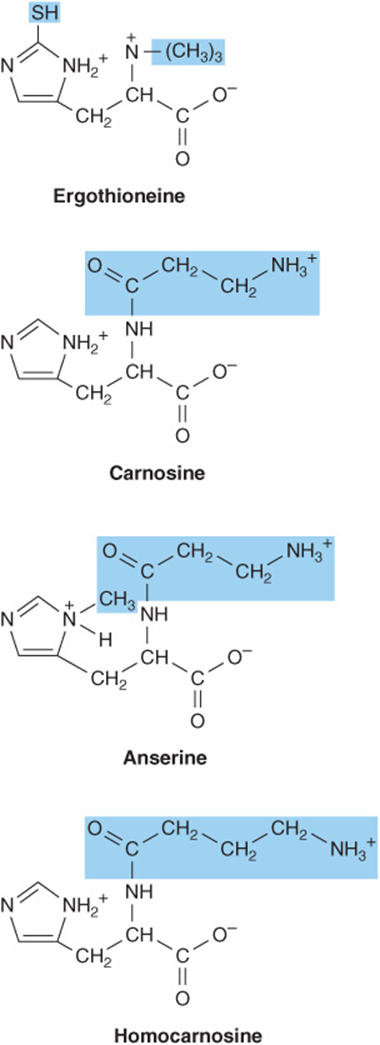
FIGURE 30–7 Derivatives of histidine. Colored boxes surround the components not derived from histidine. The SH group of ergothioneine derives from cysteine.
Methionine
The major nonprotein fate of methionine is conversion to S-adenosylmethionine, the principal source of methyl groups in the body. S-adenosylmethionine is synthesized from methionine and ATP, a reaction catalyzed by methionine adenosyltransferase (MAT) (Figure 30–8). Human tissues contain three MAT isozymes (MAT-1 and MAT-3 of liver and MAT-2 of nonhepatic tissues). Although hypermethioninemia can result from severely decreased hepatic MAT-1 and MAT-3 activity, if there is residual MAT-1 or MAT-3 activity and MAT-2 activity is normal, a high tissue concentration of methionine will assure synthesis of adequate amounts of S-adenosylmethionine.
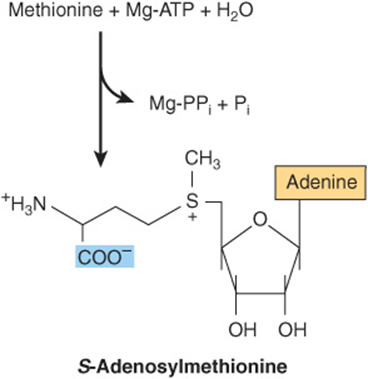
FIGURE 30–8 Biosynthesis of S-adenosylmethionine, catalyzed by methionine adenosyltransferase.
Following decarboxylation of S-adenosylmethionine by methionine decarboxylase, three carbons and the α-amino group of methionine contribute to the biosynthesis of the polyamines spermine and spermidine (Figure 30–9).These polyamines function in cell proliferation and growth, are growth factors for cultured mammalian cells, and stabilize intact cells, subcellular organelles, and membranes. Pharmacologic doses of polyamines are hypothermic and hypotensive. Since they bear multiple positive charges, polyamines associate readily with DNA and RNA. Figure 30–9 summarizes the biosynthesis of polyamines from methionine and ornithine, and Figure 30–10 the catabolism of polyamines.
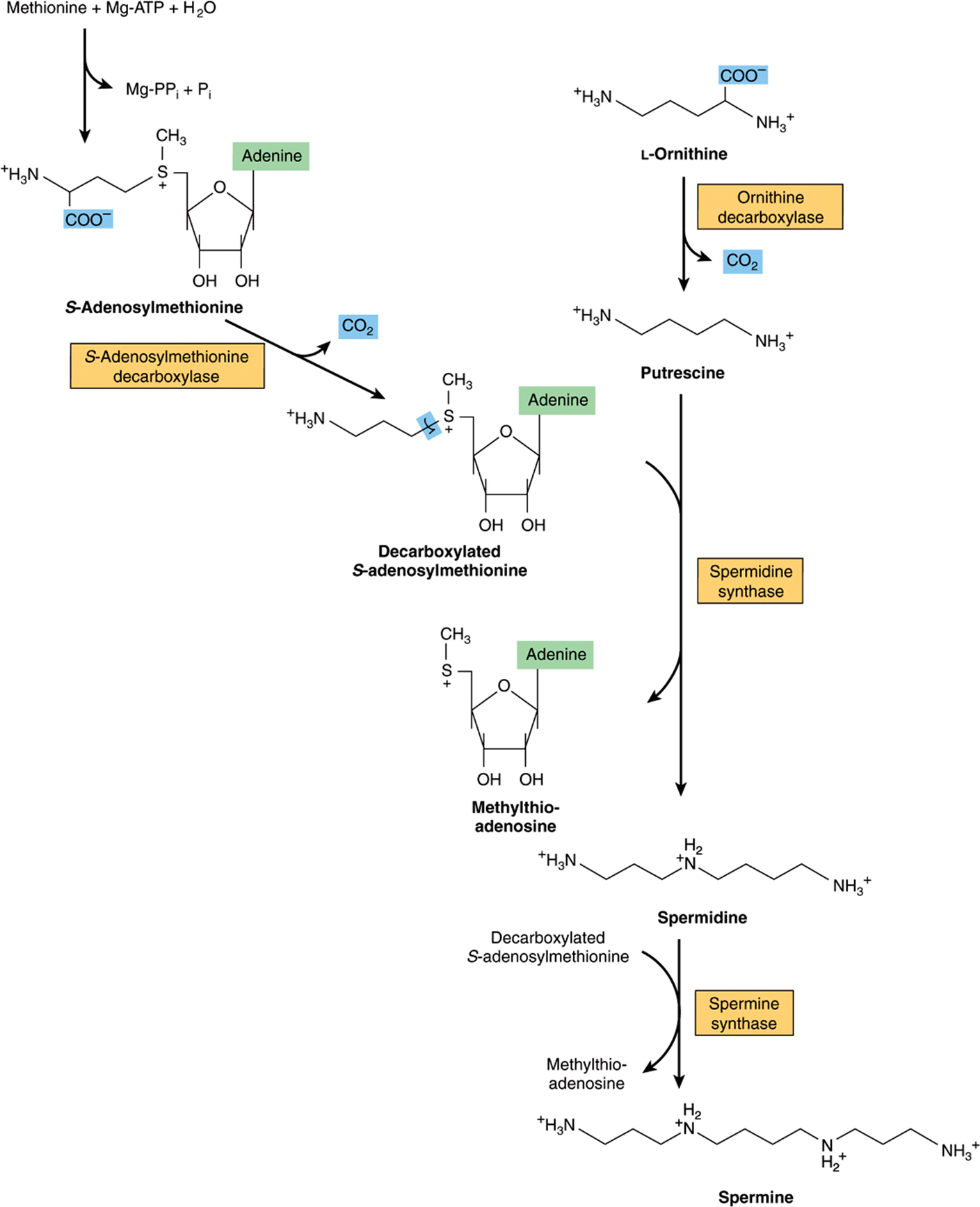
FIGURE 30–9 Intermediates and enzymes that participate in the biosynthesis of spermidine and spermine.
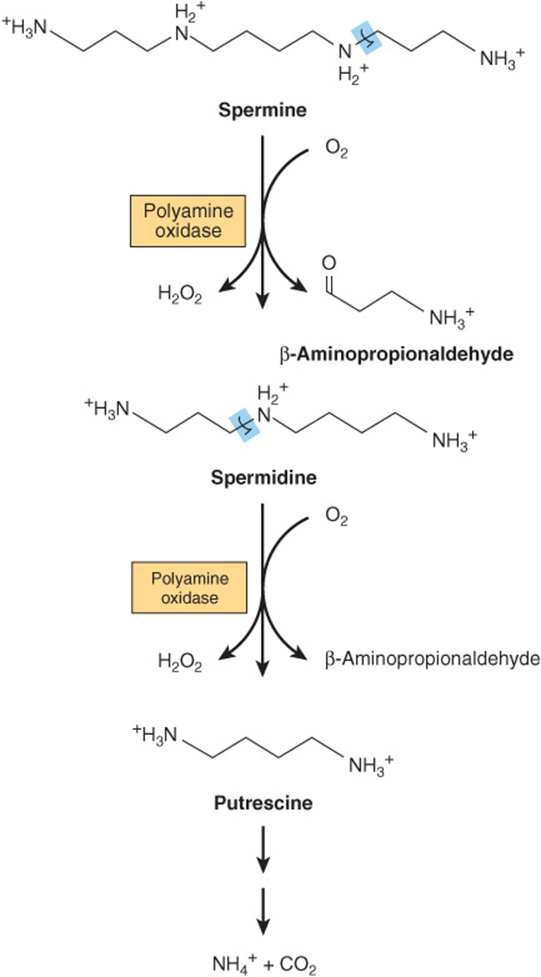
FIGURE 30–10 Catabolism of polyamines.
Serine
Serine participates in the biosynthesis of sphingosine (see Chapter 24), and of purines and pyrimidines, where it provides carbons 2 and 8 of purines and the methyl group of thymine (see Chapter 33). Genetic defects in cystathionine β-synthase, a heme protein that catalyzes the pyridoxal 5′-phosphate-dependent condensation of serine with homocysteine to form cystathionine, result in homocystinuria.
![]()
Tryptophan
Following hydroxylation of tryptophan to 5-hydroxytryptophan by liver tyrosine hydroxylase, subsequent decarboxylation forms serotonin (5-hydroxytryptamine), a potent vasoconstrictor and stimulator of smooth muscle contraction. Catabolism of serotonin is initiated by monoamine oxidase-catalyzed oxidative deamination to 5-hydroxyindole-3-acetate (Figure 30–11). The psychic stimulation that follows administration of iproniazid results from its ability to prolong the action of serotonin by inhibiting monoamine oxidase. In carcinoid (argentaffinoma), tumor cells overproduce serotonin. Urinary metabolites of serotonin in patients with carcinoid include N-acetylserotonin glucuronide and the glycine conjugate of 5-hydroxyindoleacetate. Serotonin and 5-methoxytryptamine are metabolized to the corresponding acids by monoamine oxidase. N-Acetylation of serotonin, followed by its O-methylation in the pineal body, forms melatonin. Circulating melatonin is taken up by all tissues, including brain, but is rapidly metabolized by hydroxylation followed by conjugation with sulfate or with glucuronic acid. Kidney tissue, liver tissue, and fecal bacteria all convert tryptophan to tryptamine, then to indole 3-acetate. The principal normal urinary catabolites of tryptophan are 5-hydroxyindoleacetate and indole 3-acetate.
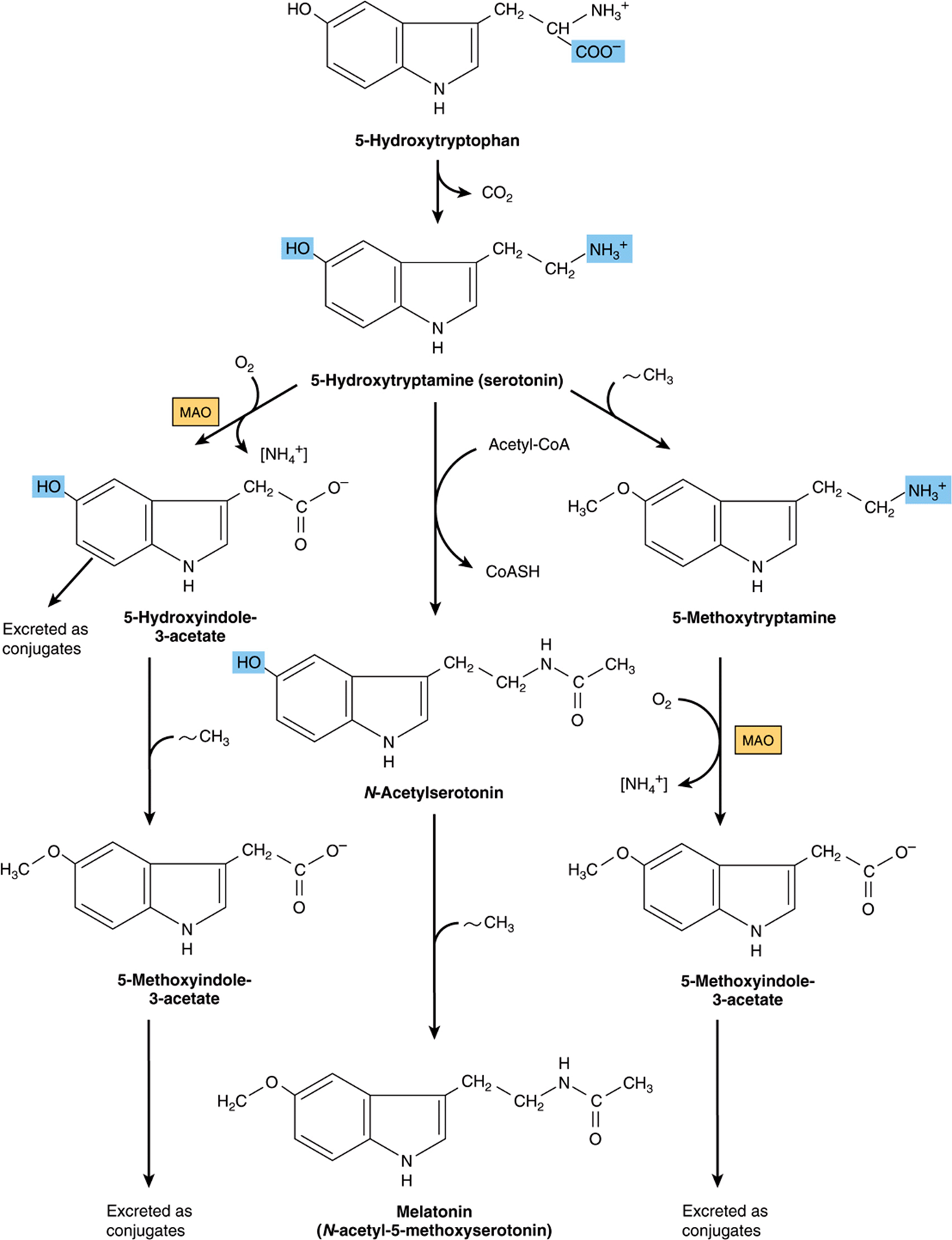
FIGURE 30–11 Biosynthesis and metabolism of serotonin and melatonin. ([NH4+], by transamination; MAO, monoamine oxidase; ~CH3, from S-adenosylmethionine.)
Tyrosine
Neural cells convert tyrosine to epinephrine and norepinephrine (Figure 30–12). While dopa is also an intermediate in the formation of melanin, different enzymes hydroxylate tyrosine in melanocytes. Dopa decarboxylase, a pyridoxal phosphate-dependent enzyme, forms dopamine. Subsequent hydroxylation by dopamine β-oxidase then forms norepinephrine. In the adrenal medulla, phenylethanolamine-N-methyltransferase utilizes S-adenosylmethionine to methylate the primary amine of norepinephrine, forming epinephrine (Figure 30–12). Tyrosine is also a precursor of triiodothyronine and thyroxine (see Chapter 41).
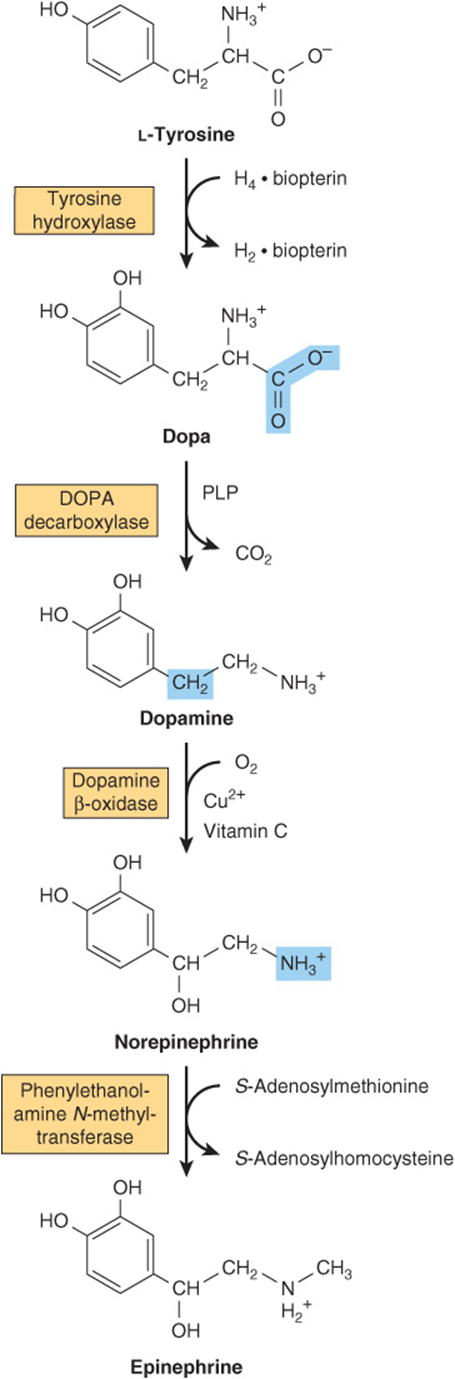
FIGURE 30–12 Conversion of tyrosine to epinephrine and norepinephrine in neuronal and adrenal cells. (PLP, pyridoxal phosphate.)
Phosphoserine, Phosphothreonine, & Phosphotyrosine
The phosphorylation and dephosphorylation of specific seryl, threonyl, or tyrosyl residues of proteins regulate the activity of certain enzymes of lipid and carbohydrate metabolism (see Chapters 9 and 19-26) and of proteins that participate in signal transduction cascades (see Chapter 42).
Sarcosine (N-Methylglycine)
The biosynthesis and catabolism of sarcosine (N-methylglycine) occur in mitochondria. Formation of sarcosine from dimethylglycine is catalyzed by the flavoprotein dimethylglycine dehydrogenase, which requires reduced pteroylpentaglutamate (TPG).
![]()
Traces of sarcosine can also arise by methylation of glycine, a reaction catalyzed by S-adenosylmethionine glycine methyltransferase.
![]()
Catabolism of sarcosine to glycine, catalyzed by the flavoprotein sarcosine dehydrogenase, also requires reduced TPG.
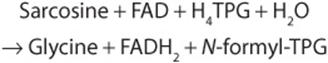
The demethylation reactions that form and degrade sarcosine represent important sources of one-carbon units. FADH2 is reoxidized via the electron transport chain (see Chapter 13).
Creatine & Creatinine
Creatinine is formed in muscle from creatine phosphate by irreversible, nonenzymatic dehydration, and loss of phosphate (Figure 30–13). Since the 24-h urinary excretion of creatinine is proportionate to muscle mass, it provides a measure of whether a complete 24-h urine specimen has been collected. Glycine, arginine, and methionine all participate in creatine biosynthesis. Synthesis of creatine is completed by methylation of guanidoacetate by S-adenosylmethionine (Figure 30–13).
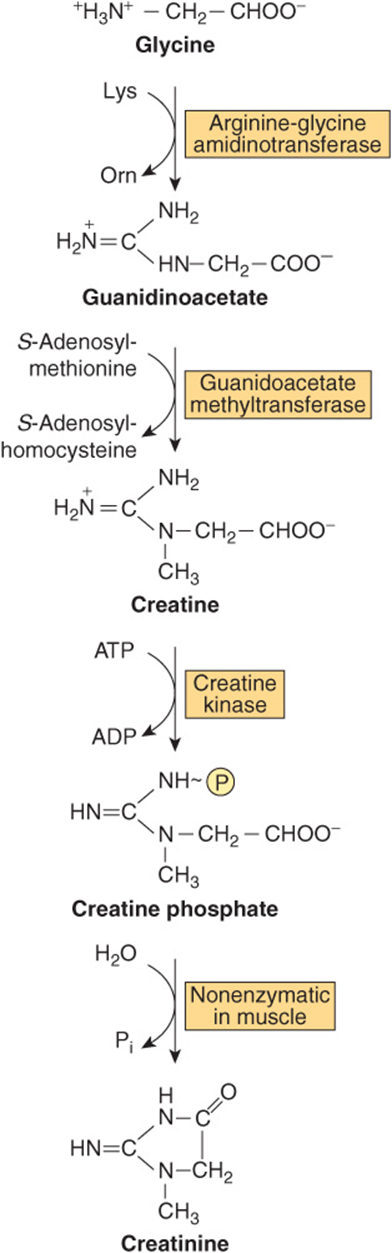
FIGURE 30–13 Biosynthesis of creatine and creatinine. Conversion of glycine and the guanidine group of arginine to creatine and creatine phosphate. Also shown is the nonenzymic hydrolysis of creatine phosphate to creatinine.
NON-α-AMINO ACIDS
Non-α-amino acids present in tissues in a free form include β-alanine, β-aminoisobutyrate, and γ-aminobutyrate (GABA). β-Alanine is also present in combined form in coenzyme A (see Figure 44–18) and in the β-alanyl dipeptides carnosine, anserine and homocarnosine (see below).
β-Alanine & β-Aminoisobutyrate
β-Alanine and β-aminoisobutyrate are formed during catabolism of the pyrimidines uracil and thymine, respectively (see Figure 33–9). Traces of β-alanine also result from the hydrolysis of β-alanyl dipeptides by the enzyme carnosinase. β-Aminoisobutyrate also arises by transamination of methylmalonate semialdehyde, a catabolite of L-valine (see Figure 29–24).
The initial reaction of β-alanine catabolism is transamination to malonate semialdehyde. Subsequent transfer of coenzyme A from succinyl-CoA forms malonyl-CoA semialdehyde, which is then oxidized to malonyl-CoA and decarboxylated to the amphibolic intermediate acetyl-CoA. Analogous reactions characterize the catabolism of β-aminoisobutyrate. Transamination forms methylmalonate semialdehyde, which is converted to the amphibolic intermediate succinyl-CoA by reactions 8V and 9V of Figure 29–24. Disorders of β-alanine and β-aminoisobutyrate metabolism arise from defects in enzymes of the pyrimidine catabolic pathway. Principal among these are disorders that result from a total or partial deficiency of dihydropyrimidine dehydrogenase (see Figure 33–9).
β-Alanyl Dipeptides
The β-alanyl dipeptides carnosine and anserine (N-methyl-carnosine) (Figure 30–7) activate myosin ATPase, chelate copper, and enhance copper uptake. β-Alanyl-imidazole buffers the pH of anaerobically contracting skeletal muscle. Biosynthesis of carnosine is catalyzed by carnosine synthetase in a two-stage reaction that involves initial formation of an enzyme-bound acyl-adenylate of β-alanine and subsequent transfer of the β-alanyl moiety to L-histidine.
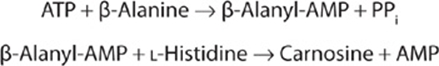
Hydrolysis of carnosine to β-alanine and L-histidine is catalyzed by carnosinase. The heritable disorder carnosinase deficiency is characterized by carnosinuria.
Homocarnosine (Figure 30–7), present in human brain at higher levels than carnosine, is synthesized in brain tissue by carnosine synthetase. Serum carnosinase does not hydrolyze homocarnosine. Homocarnosinosis, a rare genetic disorder, is associated with progressive spastic paraplegia and mental retardation.
γ-Aminobutyrate
γ-Aminobutyrate (GABA) functions in brain tissue as an inhibitory neurotransmitter by altering transmembrane potential differences. GABA is formed by decarboxylation of glutamate by L-glutamate decarboxylase (Figure 30–14).Transamination of γ-aminobutyrate forms succinate semialdehyde, which can be reduced to γ-hydroxybutyrate by L-lactate dehydrogenase, or be oxidized to succinate and thence via the citric acid cycle to CO2 and H2O (Figure 30–14). A rare genetic disorder of GABA metabolism involves a defective GABA aminotransferase, an enzyme that participates in the catabolism of GABA subsequent to its postsynaptic release in brain tissue. Defects in succinic semialdehyde dehydrogenase (Figure 30–14) are responsible for another rare metabolic disorder of γ-aminobutyrate catabolism characterized by 4-hydroxybutyric aciduria.
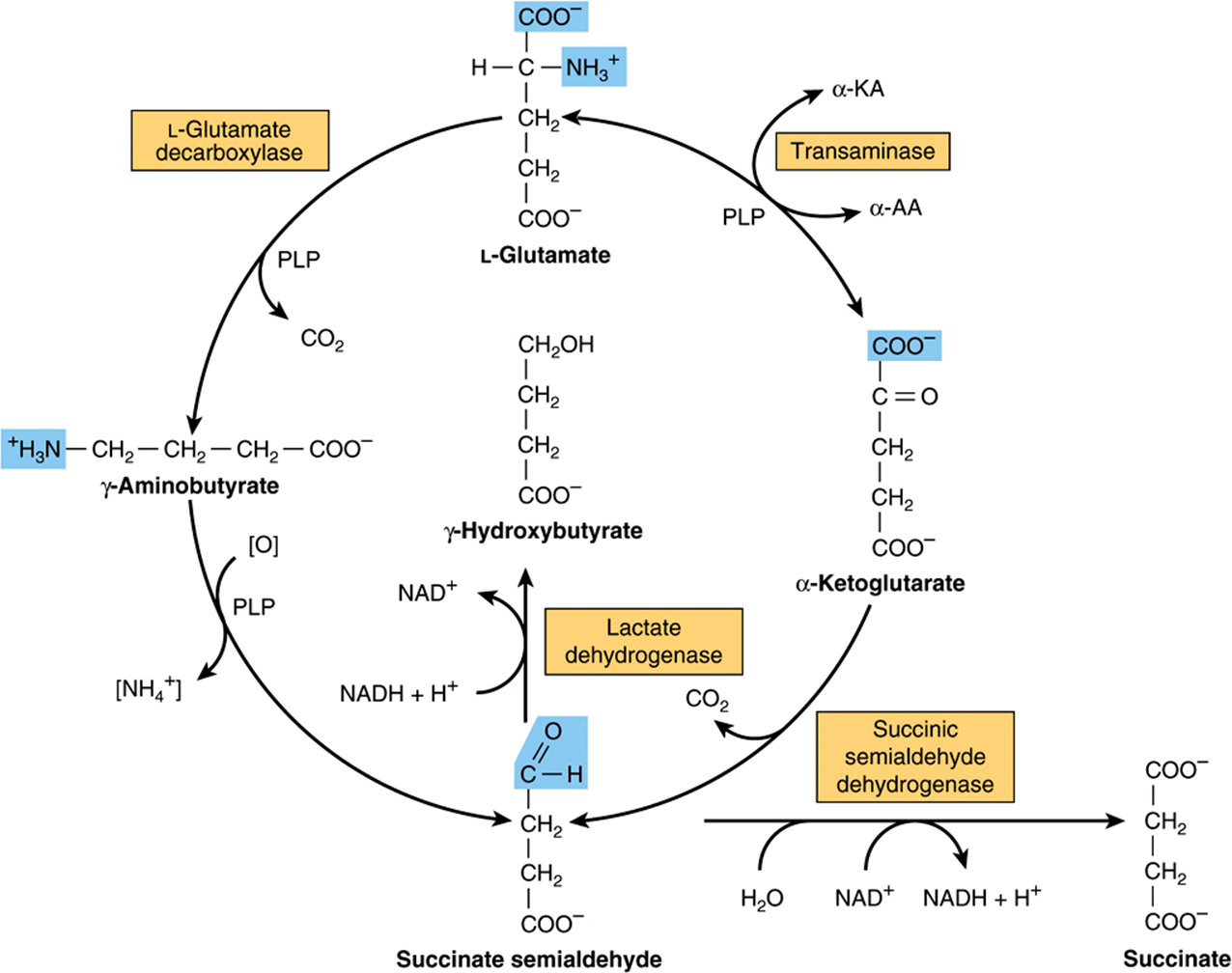
FIGURE 30–14 Metabolism of γ-aminobutyrate. (α-KA, α-keto acids; α-AA, α-amino acids; PLP, pyridoxal phosphate.)
SUMMARY
![]() In addition to serving structural and functional roles in proteins, α-amino acids participate in a wide variety of other biosynthetic processes.
In addition to serving structural and functional roles in proteins, α-amino acids participate in a wide variety of other biosynthetic processes.
![]() Arginine provides the formamidine group of creatine and the nitrogen of NO. Via ornithine, arginine provides the skeleton of the polyamines putrescine, spermine, and spermidine.
Arginine provides the formamidine group of creatine and the nitrogen of NO. Via ornithine, arginine provides the skeleton of the polyamines putrescine, spermine, and spermidine.
![]() Cysteine provides the thioethanolamine portion of coenzyme A, and following its conversion to taurine, part of the bile acid taurocholic acid.
Cysteine provides the thioethanolamine portion of coenzyme A, and following its conversion to taurine, part of the bile acid taurocholic acid.
![]() Glycine participates in the biosynthesis of heme, purines, creatine, and N-methylglycine (sarcosine). Many drugs and drug metabolites are excreted as glycine conjugates, which increases water solubility for urinary excretion.
Glycine participates in the biosynthesis of heme, purines, creatine, and N-methylglycine (sarcosine). Many drugs and drug metabolites are excreted as glycine conjugates, which increases water solubility for urinary excretion.
![]() Decarboxylation of histidine forms the neurotransmitter histamine. Histidine compounds present in the human body include ergothioneine, carnosine, and dietary anserine.
Decarboxylation of histidine forms the neurotransmitter histamine. Histidine compounds present in the human body include ergothioneine, carnosine, and dietary anserine.
![]() S-Adenosylmethionine, the principal source of methyl groups in metabolism, contributes its carbon skeleton to the biosynthesis of the polyamines spermine and spermidine.
S-Adenosylmethionine, the principal source of methyl groups in metabolism, contributes its carbon skeleton to the biosynthesis of the polyamines spermine and spermidine.
![]() In addition to its roles in phospholipid and sphingosine biosynthesis, serine provides carbons 2 and 8 of purines and the methyl group of thymine.
In addition to its roles in phospholipid and sphingosine biosynthesis, serine provides carbons 2 and 8 of purines and the methyl group of thymine.
![]() Key tryptophan metabolites include serotonin and melatonin. Kidney and liver tissue, and also fecal bacteria, convert tryptophan to tryptamine and thence to indole 3-acetate, The principal tryptophan catabolites in urine are indole 3-acetate and 5-hydroxyindoleacetate.
Key tryptophan metabolites include serotonin and melatonin. Kidney and liver tissue, and also fecal bacteria, convert tryptophan to tryptamine and thence to indole 3-acetate, The principal tryptophan catabolites in urine are indole 3-acetate and 5-hydroxyindoleacetate.
![]() Tyrosine forms norepinephrine and epinephrine, and following iodination the thyroid hormones triiodothyronine and thyroxine.
Tyrosine forms norepinephrine and epinephrine, and following iodination the thyroid hormones triiodothyronine and thyroxine.
![]() The enzyme-catalyzed interconversion of the phospho- and dephospho-forms of peptide bound serine, threonine, and tyrosine plays key roles in metabolic regulation, including signal transduction.
The enzyme-catalyzed interconversion of the phospho- and dephospho-forms of peptide bound serine, threonine, and tyrosine plays key roles in metabolic regulation, including signal transduction.
![]() Glycine, arginine, and S-adenosylmethionine all participate in the biosynthesis of creatine, which as creatine phosphate serves as a major energy reserve in muscle and brain tissue. Excretion in the urine of its catabolite creatinine is proportionate to muscle mass.
Glycine, arginine, and S-adenosylmethionine all participate in the biosynthesis of creatine, which as creatine phosphate serves as a major energy reserve in muscle and brain tissue. Excretion in the urine of its catabolite creatinine is proportionate to muscle mass.
![]() β-Alanine and β-aminoisobutyrate both are present in tissues as free amino acids. β-Alanine also occurs in bound form in coenzyme A, carnosine, anserine, and homocarnosine. Catabolism of β-alanine involves stepwise conversion to acetyl-CoA. Analogous reactions catabolize β-aminoisobutyrate to succinyl-CoA. Disorders of β-alanine and β-aminoisobutyrate metabolism arise from defects in enzymes of pyrimidine catabolism.
β-Alanine and β-aminoisobutyrate both are present in tissues as free amino acids. β-Alanine also occurs in bound form in coenzyme A, carnosine, anserine, and homocarnosine. Catabolism of β-alanine involves stepwise conversion to acetyl-CoA. Analogous reactions catabolize β-aminoisobutyrate to succinyl-CoA. Disorders of β-alanine and β-aminoisobutyrate metabolism arise from defects in enzymes of pyrimidine catabolism.
![]() Decarboxylation of glutamate forms the inhibitory neurotransmitter γ-aminobutyrate (GABA). Two rare metabolic disorders are associated with defects in GABA catabolism.
Decarboxylation of glutamate forms the inhibitory neurotransmitter γ-aminobutyrate (GABA). Two rare metabolic disorders are associated with defects in GABA catabolism.
REFERENCES
Conti M, Beavo J: Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 2007;76:481.
Dominy JE Jr, Hwang J, Guo S, et al: Synthesis of cysteine dioxygenase’s amino acid cofactor is regulated by substrate and represents a novel post-translational regulation of activity. J Biol Chem 2008;283:12188.
Joseph CA, Maroney MJ: Cysteine dioxygenase: structure and mechanism. Chem Commun (Camb) 2007;28:3338.
Lindemose S, Nielsen PE, Mollegaard NE: Polyamines preferentially interact with bent adenine tracts in double-stranded DNA. Nucleic Acids Res 2005;33:1790.
Manegold C, Hoffmann GF, Degen I, et al: Aromatic L-amino acid decarboxylase deficiency: clinical features, drug therapy and followup. J Inherit Metab Dis 2009;32:371.
Moinard C, Cynober L, de Bandt JP: Polyamines: metabolism and implications in human diseases. Clin Nutr 2005;24:184.
Pearl PL, Gibson KM, Cortez MA, et al: Succinic semialdehyde dehydrogenase deficiency: Lessons from mice and men. J Inherit Metab Dis 2009;32:343.
Pearl PL, Taylor JL, Trzcinski S, et al: The pediatric neurotransmitter disorders. J Child Neurol 2007;22:606.
Scriver CR, Sly WS, Childs B, et al (editors): The Metabolic and Molecular Bases of Inherited Disease, 8th ed. McGraw-Hill, 2001.
Wu F, Yu J, Gehring H. Inhibitory and structural studies of novel coenzyme-substrate analogs of human histidine decarboxylase. FASEB J. 2008;3:890.