Harper’s Illustrated Biochemistry, 29th Edition (2012)
SECTION VI. Special Topics
Chapter 46. Intracellular Traffic & Sorting of Proteins
Robert K. Murray, MD, PhD
OBJECTIVES
After studying this chapter, you should be able to:
![]() Know that many proteins are targeted by signal sequences to their correct destinations and that the Golgi apparatus plays an important role in sorting proteins.
Know that many proteins are targeted by signal sequences to their correct destinations and that the Golgi apparatus plays an important role in sorting proteins.
![]() Understand that specialized signals are involved in sorting proteins to mitochondria, the nucleus, and to peroxisomes.
Understand that specialized signals are involved in sorting proteins to mitochondria, the nucleus, and to peroxisomes.
![]() Appreciate that N-terminal signal peptides play a key role in directing newly synthesized proteins into the lumen of the endoplasmic reticulum.
Appreciate that N-terminal signal peptides play a key role in directing newly synthesized proteins into the lumen of the endoplasmic reticulum.
![]() Know that chaperones prevent faulty folding of other proteins, that mechanisms exist for disposing of misfolded proteins, and that the endoplasmic reticulum acts as a quality control compartment.
Know that chaperones prevent faulty folding of other proteins, that mechanisms exist for disposing of misfolded proteins, and that the endoplasmic reticulum acts as a quality control compartment.
![]() Comprehend that ubiquitin is a key molecule in protein degradation.
Comprehend that ubiquitin is a key molecule in protein degradation.
![]() Recognize the important role of transport vesicles in intracellular transport.
Recognize the important role of transport vesicles in intracellular transport.
![]() Appreciate that many diseases result from mutations in genes encoding proteins involved in intracellular transport and be familiar with the terms conformational diseases and diseases of proteostatic deficiency.
Appreciate that many diseases result from mutations in genes encoding proteins involved in intracellular transport and be familiar with the terms conformational diseases and diseases of proteostatic deficiency.
BIOMEDICAL IMPORTANCE
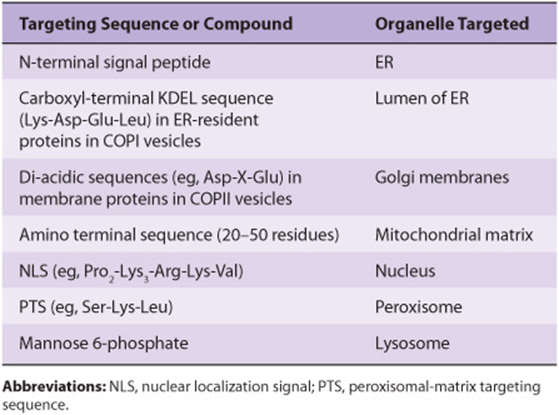
Proteins must travel from polyribosomes, where they are synthesized, to many different sites in the cell to perform their particular functions. Some are destined to be components of specific organelles, others for the cytosol or for export, and yet others will be located in the various cellular membranes. Thus, there is considerable intracellular traffic of proteins. A major insight was the recognition by Blobel and subsequently others that for proteins to attain their proper locations, they generally contain information (a signal or coding sequence) that targets them appropriately. Once a number of the signals were defined (see Table 46-1), it became apparent that certain diseases result from mutations that affect these signals. In this chapter, we discuss the intracellular traffic of proteins and their sorting and briefly consider some of the disorders that result when abnormalities occur.
TABLE 46–1 Some Sequences or Molecules That Direct Proteins to Specific Organelles
MANY PROTEINS ARE TARGETED BY SIGNAL SEQUENCES TO THEIR CORRECT DESTINATIONS
The protein biosynthetic pathways in cells can be considered to be one large sorting system. Many proteins carry signals (usually but not always specific sequences of amino acids) that direct them to their destination, thus ensuring that they will end up in the appropriate membrane or cell compartment; these signals are a fundamental component of the sorting system. Usually, the signal sequences are recognized and interact with complementary areas of other proteins that serve as receptors for those containing the signals.
A major sorting decision is made early in protein biosynthesis, when specific proteins are synthesized either on free or on membrane-bound polyribosomes. It is important to understand that these two types of ribosomes are identical in structure and potentially interchangeable. However, if a growing polypeptide chain attached to polyribosomes lacks an N-terminal signal peptide (see below), it will not interact with the membrane of the endoplasmic reticulum (ER) and the polyribosomes are described as cytosolic. On the other hand, a growing polypeptide chain that contains an N-terminal signal peptide will interact with the membrane of the ER, and the polyribosomes to which it is attached are described as membrane-bound. This results in two sorting branches, called the cytosolic branch and the rough ER (RER) branch (Figure 46–1).
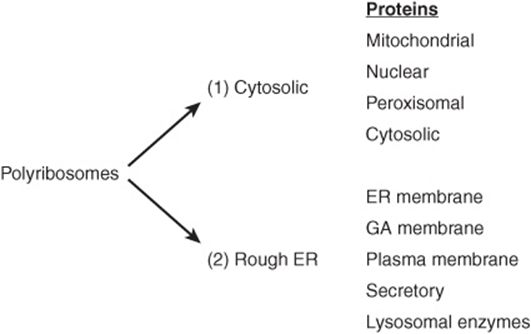
FIGURE 46–1 Diagrammatic representation of the two branches of protein sorting occurring by synthesis on (1) cytosolic and (2) membrane-bound polyribosomes. The mitochondrial proteins listed are encoded by nuclear genes; one of the signals used in further sorting of mitochondrial matrix proteins is listed in Table 46-1. (ER, endoplasmic reticulum; GA, Golgi apparatus.)
Proteins synthesized by cytosolic polyribosomes are directed to mitochondria, nuclei, and peroxisomes by specific signals, or remain in the cytosol if they lack a signal. Any protein that contains a targeting sequence that is subsequently removed is designated as a preprotein. In some cases, a second peptide is also removed, and in that event the original protein is known as a preproprotein (eg, preproalbumin; Chapter 50).
Proteins synthesized and sorted in the rough ER branch (Figure 46–1) include many destined for various membranes (eg, of the ER, Golgi apparatus [GA], plasma membrane [PM]) and for secretion. Lysosomal enzymes are also included. These various proteins may thus reside in the membranes or lumen of the ER, or follow the major transport route of intracellular proteins to the GA. The entire pathway of ER—GA—plasma membrane is often called the secretory or exocytotic pathway. The secretory pathway was first delineated by the work of George Palade and colleagues, using radioactive amino acids and radioautography to follow the fate of proteins synthesized in the exocrine pancreas. Events along the secretory pathway will be given special attention here. Proteins destined for the GA, the PM, certain other sites, or for secretion are carried in transport vesicles (Figure 46–2); a brief description of the formation of these important particles will be given subsequently. Certain other proteins destined for secretion are carried in secretory vesicles (Figure 46–2). These are prominent in the pancreas and certain other glands. Their mobilization and discharge are regulated and often referred to as “regulated secretion”. In contrast, transport of vesicles occurring continuously through the secretory pathway is referred to as “constitutive transport”. Passage of enzymes to the lysosomes using the mannose 6-phosphate signal is described in Chapter 47.
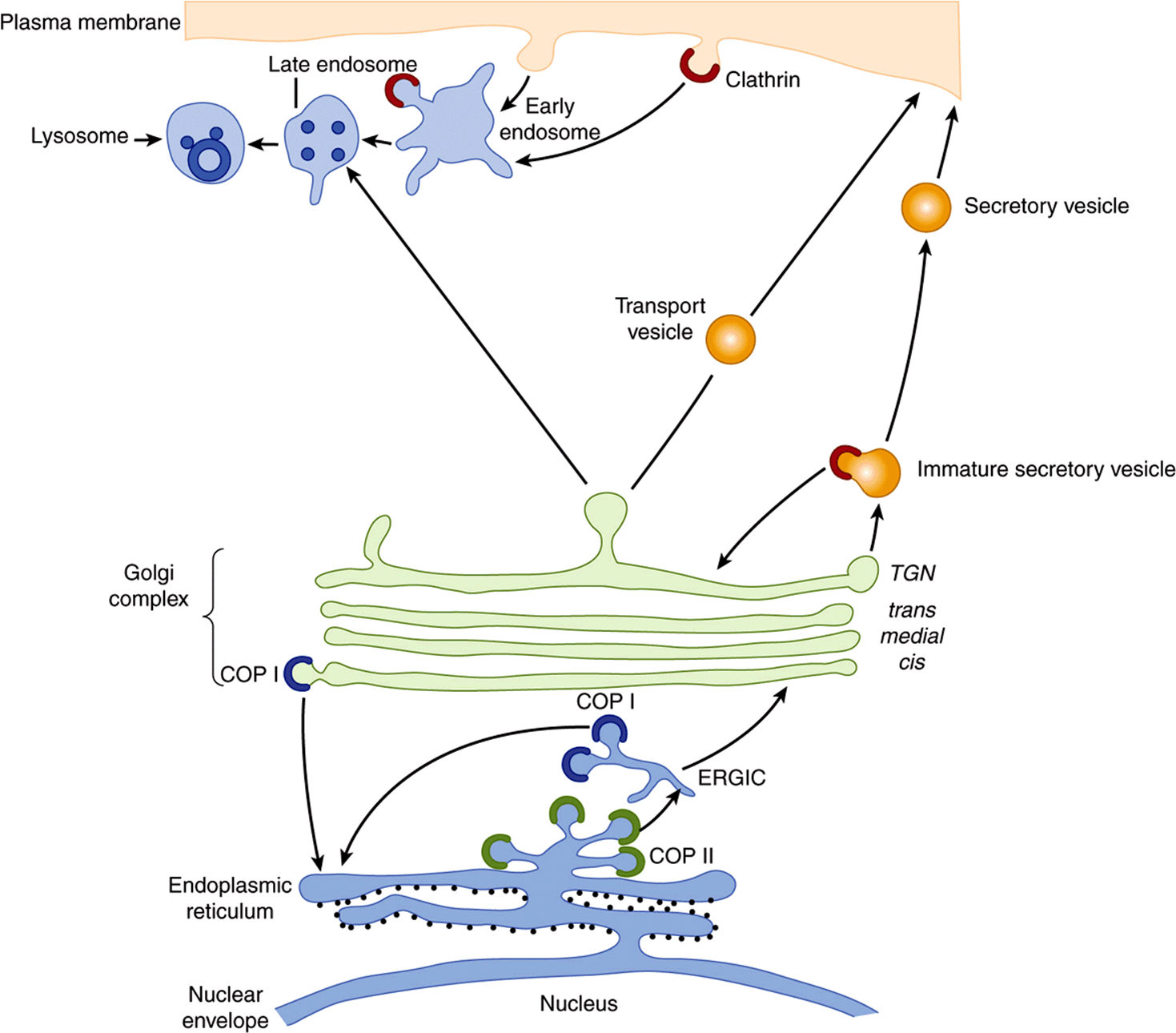
FIGURE 46–2 Diagrammatic representation of the rough ER branch of protein sorting. Newly synthesized proteins are inserted into the ER membrane or lumen from membrane-bound polyribosomes (small black circles studding the cytosolic face of the ER). Proteins that are transported out of the ER are carried in COPII vesicles to the cis-Golgi (anterograde transport). Movement of proteins through the Golgi appears to be mainly by cisternal maturation. In the TGN, the exit side of the Golgi, proteins are segregated and sorted. Secretory proteins accumulate in secretory vesicles (regulated secretion), from which they are expelled at the plasma membrane. Proteins destined for the plasma membrane or those that are secreted in a constitutive manner are carried out to the cell surface in transport vesicles (constitutive secretion). Clathrin-coated vesicles are involved in endocytosis, carrying cargo to late endosomes and to lysosomes. Mannose 6-phosphate (not shown; see Chapter 47) acts as a signal for transporting enzymes to lysosomes. COPI vesicles are involved in retrieving proteins from the Golgi to the ER (retrograde transport) and may be involved in some intra-Golgi transport. COP II vesicles are involved in concentrating cargo for export from the ER to the GA. Cargo normally passes through the ERGIC compartment to the GA. (TGN, trans-Golgi network; ERGIC, ER-Golgi intermediate complex.) (Courtesy of E Degen.)
The Golgi Apparatus Is Involved in Glycosylation & Sorting of Proteins
The GA plays two major roles in protein synthesis. First, it is involved in the processing of the oligosaccharide chains of membrane and other N-linked glycoproteins and also contains enzymes involved in O-glycosylation (see Chapter 47). Second, it is involved in the sorting of various proteins prior to their delivery to their appropriate intracellular destinations. All parts of the GA participate in the first role, whereas the frans-Golgi network (TGN) is particularly involved in the second and is very rich in vesicles.
A Wide Variety of Experimental Techniques Have Been Used to Investigate Trafficking and Sorting
Approaches that have afforded major insights to the processes described in this chapter include (1) electron microscopy; (2) use of yeast mutants; (3) subcellular fractionation; (4) application of recombinant DNA techniques (eg, mutating or eliminating particular sequences in proteins, or fusing new sequences onto them); and (5) development of in vitro systems (eg, to study translocation into the ER and mechanisms of vesicle formation); (6) use of fluorescent tags to follow the movement of proteins; and (7) structural studies on certain proteins, particularly by x-ray crystallography.
The sorting of proteins belonging to the cytosolic branch referred to above is described next, starting with mitochondrial proteins.
THE MITOCHONDRION BOTH IMPORTS & SYNTHESIZES PROTEINS
Mitochondria contain many proteins. Thirteen polypeptides (mostly membrane components of the electron transport chain) are encoded by the mitochondrial (mt) genome and synthesized in that organelle using its own protein-synthesizing system. However, the majority (at least several hundred) are encoded by nuclear genes, are synthesized outside the mitochondria on cytosolic polyribosomes, and must be imported. Yeast cells have proved to be a particularly useful system for analyzing the mechanisms of import of mitochondrial proteins, partly because it has proved possible to generate a variety of mutants that have illuminated the fundamental processes involved. Most progress has been made in the study of proteins present in the mitochondrial matrix, such as the F1 ATPase subunits. Only the pathway of import of matrix proteins will be discussed in any detail here.
Matrix proteins must pass from cytosolic polyribosomes through the outer and inner mitochondrial membranes to reach their destination. Passage through the two membranes is called translocation. They have an amino terminal leader sequence (presequence), about 20-50 amino acids in length (see Table 46-1), which is not highly conserved but is amphipathic and contains many hydrophobic and positively charged amino acids (eg, Lys or Arg). The presequence is equivalent to a signal peptide mediating attachment of polyribosomes to membranes of the ER (see below), but in this instance targeting proteins to the matrix. Some general features of the passage of a protein from the cytosol to the mt matrix are shown in Figure 46–3.
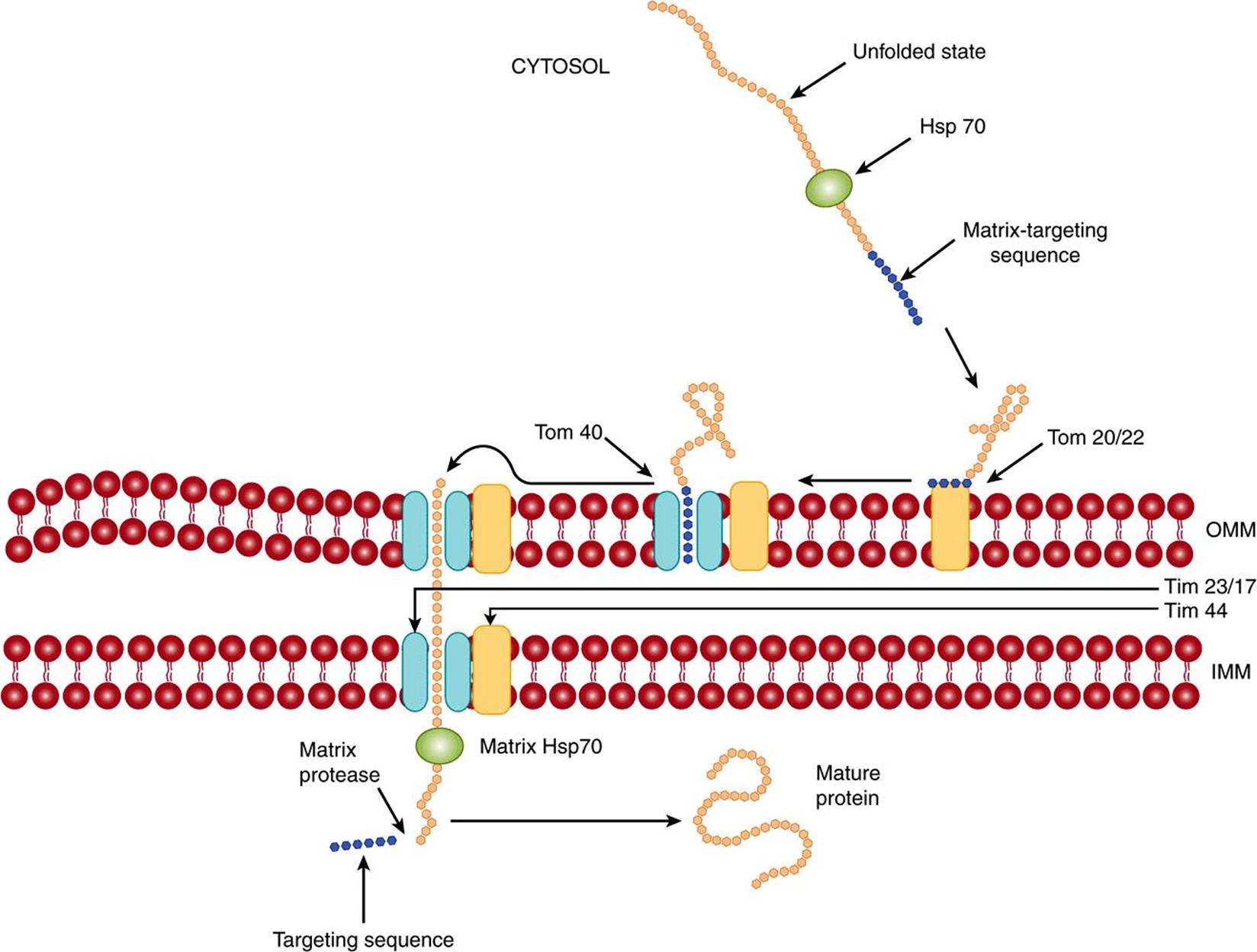
FIGURE 46–3 Schematic representation of the entry of a protein into the mitochondrial matrix. The unfolded protein synthesized on cytosolic poyribosomes and containing a matrix-targeting sequence interacts with the cytosolic chaperone Hsp 70. The protein next interacts with the mt outer membrane receptor Tom 20/22, and is transferred to the neighboring import channel Tom 40 (Tom, translocon of the outer membrane). The protein is then translocated across the channel; the channel on the inner mt membrane is largely composed of Tim 23 and Tim 17 proteins (Tim, translocon of the inner membrane). On the inside of the inner mt membrane, it interacts with the matrix chaperone Hsp 70, which in turn interacts with membrane protein Tim 44. The hydrolysis of ATP by mt Hsp70 probably helps drive the translocation, as does the electronegative interior of the matrix. The targeting sequence is subsequently cleaved by the matrix processing enzyme, and the imported protein assumes its final shape, or may interact with an mt chaperonin prior to this. At the site of translocation, the outer and inner mt membranes are in close contact. OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane. (Modified, with permission, from Lodish H, et al: Molecular Cell Biology, 6th ed. W.H. Freeman & Co., 2008.)
Translocation occurs posttranslationally, after the matrix proteins are released from the cytosolic polyribosomes. Interactions with a number of cytosolic proteins that act as chaperones (see below) and as targeting factorsoccur prior to translocation.
Two distinct translocation complexes are situated in the outer and inner mitochondrial membranes, referred to (respectively) as TOM (translocase-of-the-outer membrane) and TIM (translocase-of-the-inner membrane). Each complex has been analyzed and found to be composed of a number of proteins, some of which act as receptors (eg, Tom20/22) for the incoming proteins and others as components (eg, Tom40) of the transmembrane poresthrough which these proteins must pass. Proteins must be in the unfolded state to pass through the complexes, and this is made possible by ATP-dependent binding to several chaperone proteins. The roles of chaperone proteins in protein folding are discussed later in this chapter. In mitochondria, they are involved in translocation, sorting, folding, assembly, and degradation of imported proteins. A proton-motive force across the inner membrane is required for import; it is made up of the electric potential across the membrane (inside negative) and the pH gradient (see Chapter 13). The positively charged leader sequence may be helped through the membrane by the negative charge in the matrix. The presequence is split off in the matrix by a matrix-processing protease (MPP). Contact with other chaperones present in the matrix is essential to complete the overall process of import. Interaction with mt-Hsp70 (mt = mitochondrial; Hsp = heat shock protein; ![]() kDa) ensures proper import into the matrix and prevents misfolding or aggregation, while interaction with the mt-Hsp60-Hsp10 system ensures proper folding. The interactions of imported proteins with the above chaperones require hydrolysis of ATP to drive them.
kDa) ensures proper import into the matrix and prevents misfolding or aggregation, while interaction with the mt-Hsp60-Hsp10 system ensures proper folding. The interactions of imported proteins with the above chaperones require hydrolysis of ATP to drive them.
The details of how preproteins are translocated have not been fully elucidated. It is possible that the electric potential associated with the inner mitochondrial membrane causes a conformational change in the unfolded preprotein being translocated and that this helps to pull it across. Furthermore, the fact that the matrix is more negative than the intermembrane space may “attract” the positively charged amino terminal of the preprotein to enter the matrix. Close apposition at contact sites between the outer and inner membranes is necessary for translocation to occur.
The above describes the major pathway of proteins destined for the mitochondrial matrix. However, certain proteins insert into the outer mitochondrial membrane facilitated by the TOM complex. Others stop in the intermembrane space, and some insert into the inner membrane. Yet others proceed into the matrix and then return to the inner membrane or intermembrane space. A number of proteins contain two signaling sequences—one to enter the mitochondrial matrix and the other to mediate subsequent relocation (eg, into the inner membrane). Certain mitochondrial proteins do not contain presequences (eg, cytochrome c, which locates in the inter membrane space), and others contain internal presequences. Overall, proteins employ a variety of mechanisms and routes to attain their final destinations in mitochondria.
General features that apply to the import of proteins into organelles, including mito chondria and some of the other organelles to be discussed below, are summarized in Table 46-2.
TABLE 46–2 Some General Features of Protein Import to Organelles

LOCALIZATION SIGNALS, IMPORTINS, & EXPORTINS ARE INVOLVED IN TRANSPORT OF MACROMOLECULES IN & OUT OF THE NUCLEUS
It has been estimated that more than a million macromolecules per minute are transported between the nucleus and the cytoplasm in an active eukaryotic cell. These macromolecules include histones, ribosomal proteins and ribosomal subunits, transcription factors, and mRNA molecules. The transport is bidirectional and occurs through the nuclear pore complexes (NPCs). These are complex structures with a mass approximately 15 times that of a ribosome and are composed of aggregates of about 30 different proteins. The minimal diameter of an NPC is approximately 9 nm. Molecules smaller than about 40 kDa can pass through the channel of the NPC by diffusion, but special translocation mechanisms exist for larger molecules. These mechanisms are under intensive investigation, but some important features have already emerged.
Here we shall mainly describe nuclear import of certain macromolecules. The general picture that has emerged is that proteins to be imported (cargo molecules) carry a nuclear localization signal (NLS). One example of an NLS is the amino acid sequence (Pro)2-(Lys)3-Arg-Lys-Val (see Table 46-1), which is markedly rich in basic residues. Depending on which NLS it contains, a cargo molecule interacts with one of a family of soluble proteins called importins, and the complex docks transiently at the NPC. Another family of proteins called Ran plays a critical regulatory role in the interaction of the complex with the NPC and in its translocation through the NPC. Ran proteins are small monomeric nuclear GTPases and, like other GTPases, exist in either GTP-bound or GDP-bound states. They are themselves regulated by guanine nucleotide exchange factors (GEFs), which are located in the nucleus, and Ran GTPase-accelerating proteins (GAPs), which are predominantly cytoplasmic. The GTP-bound state of Ran is favored in the nucleus and the GDP-bound state in the cytoplasm. The conformations and activities of Ran molecules vary depending on whether GTP or GDP is bound to them (the GTP-bound state is active; see discussion of G proteins in Chapter 42). The asymmetry between nucleus and cytoplasm—with respect to which of these two nucleotides is bound to Ran molecules—is thought to be crucial in understanding the roles of Ran in transferring complexes unidirectionally across the NPC. When cargo molecules are released inside the nucleus, the importins recirculate to the cytoplasm to be used again. Figure 46–4 summarizes some of the principal features in the above process.
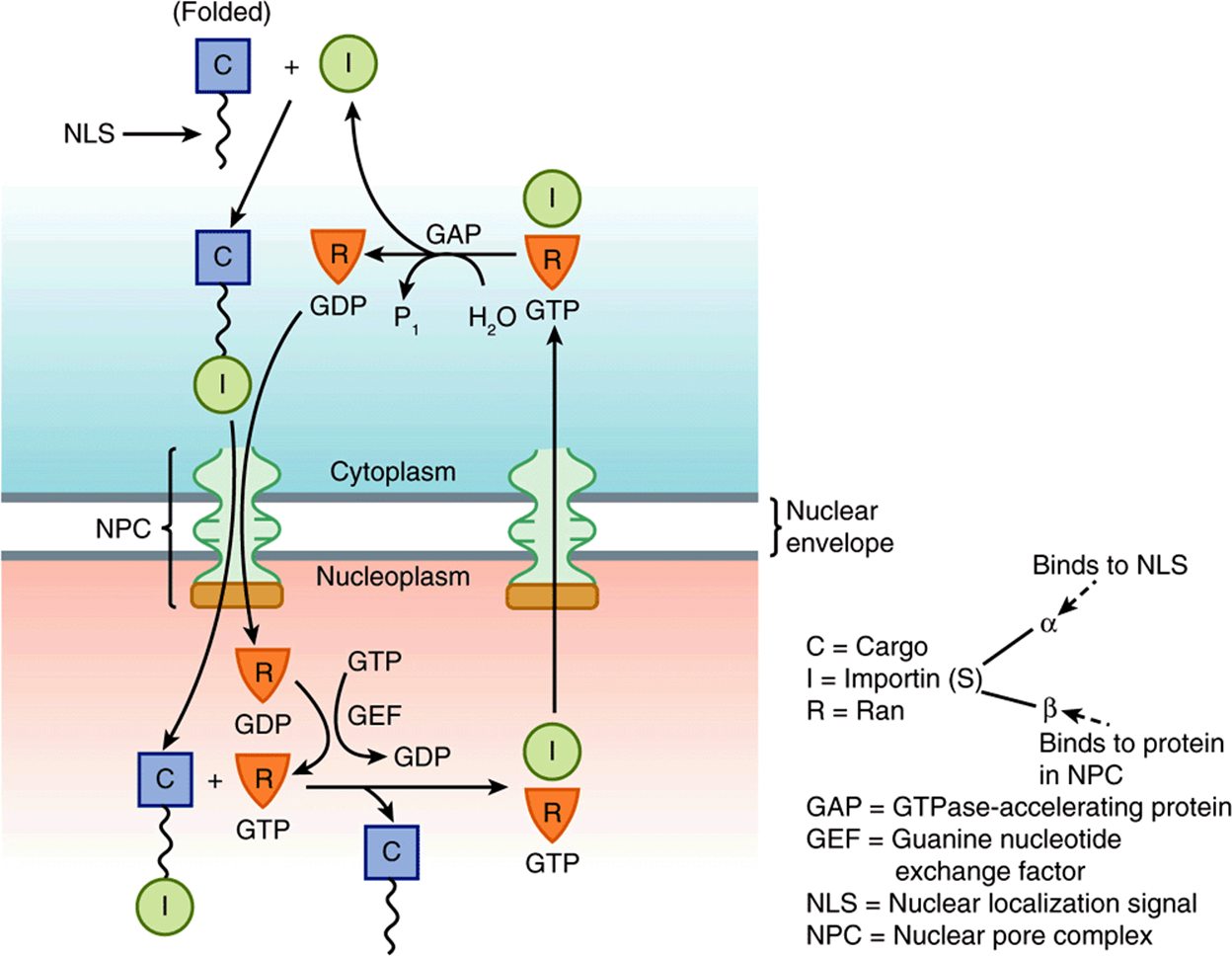
FIGURE 46–4 Simplified representation of the entry of a protein into the nucleoplasm. As shown in the top left-hand side of the figure, a cargo molecule in the cytoplasm via its NLS interacts to form a complex with an importin. (This may be either importin α or both importin α and importin β). This complex next interacts with Ran-GDP and traverses the NPC into the nucleoplasm. In the nucleoplasm, Ran-GDP is converted to Ran-GTP by GEF, causing a conformational change in Ran resulting in the cargo molecule being released. The importin-Ran-GTP complex then leaves the nucleoplasm via the NPC to return to the cytoplasm. In the cytoplasm, due to the action of GTPase-accelerating protein (GAP), which converts GTP to GDP, the importin is released to participate in another import cycle. The Ran-GTP is the active form of the complex, with the Ran-GDP form being considered inactive. Directionality is believed to be conferred on the overall process by the dissociation of Ran-GTP in the cytoplasm. (C, cargo molecule; GAP, GTPase-accelerating protein; GEF, guanine nucleotide exchange factor; I, importin; NLS, nuclear localizing signal; NPC, nuclear pore complex.) (Modified, with permission, from Lodish H, et al: Molecular Cell Biology, 6th ed. W.H. Freeman & Co., 2008.)
Proteins similar to importins, referred to as exportins, are involved in the export of many macromolecules (various proteins, tRNA molecules, ribosomal subunits and certain mRNA molecules) from the nucleus. Cargo molecules for export carry nuclear export signals (NESs). Ran proteins are involved in this process also, and it is now established that the processes of import and export share a number of common features. The family of importins and exportins are referred to as karyopherins.
Another system is involved in the translocation of the majority of mRNA molecules. These are exported from the nucleus to the cytoplasm as ribonucleoprotein (RNP) complexes attached to a protein named mRNP exporter.This is a heterodimeric molecule (ie, composed of two different subunits, TAP and Nxt-1) that carries RNP molecules through the NPC. Ran is not involved. This system appears to use the hydrolysis of ATP by an RNA helicase (Dbp5) to drive translocation.
Other small monomeric GTPases (eg, ARF, Rab, Ras, and Rho) are important in various cellular processes such as vesicle formation and transport (ARF and Rab; see below), certain growth and differentiation processes (Ras), and formation of the actin cytoskeleton (Rho). A process involving GTP and GDP is also crucial in the transport of proteins across the membrane of the ER (see below).
PROTEINS IMPORTED INTO PEROXISOMES CARRY UNIQUE TARGETING SEQUENCES
The peroxisome is an important organelle involved in aspects of the metabolism of many molecules, including fatty acids and other lipids (eg, plasmalogens, cholesterol, bile acids), purines, amino acids, and hydrogen peroxide. The peroxisome is bounded by a single membrane and contains more than 50 enzymes; catalase and urate oxidase are marker enzymes for this organelle. Its proteins are synthesized on cytosolic polyribosomes and fold prior to import. The pathways of import of a number of its proteins and enzymes have been studied, some being matrix components (see Figure 46–5) and others membrane components. At least two peroxisomal-matrix targeting sequences (PTSs) have been discovered. One, PTS1, is a tripeptide (ie, Ser-Lys-Leu [SKL], but variations of this sequence have been detected) located at the carboxyl terminal of a number of matrix proteins, including catalase. Another, PTS2, is at the N-terminus and has been found in at least four matrix proteins (eg, thiolase). Neither of these two sequences is cleaved after entry into the matrix. Proteins containing PTS1 sequences form complexes with a cytosolic receptor protein (Pex5) and proteins containing PTS2 sequences complex with another receptor protein. The resulting complexes then interact with a membrane receptor complex, Pex2/10/12, which translocates them into the matrix. Proteins involved in further transport of proteins into the matrix are also present. Pex5 is recycled to the cytosol. Most peroxisomal membrane proteins have been found to contain neither of the above two targeting sequences, but apparently contain others. The import system can handle intact oligomers (eg, tetrameric catalase). Import of matrix proteins requires ATP, whereas import of membrane proteins does not.
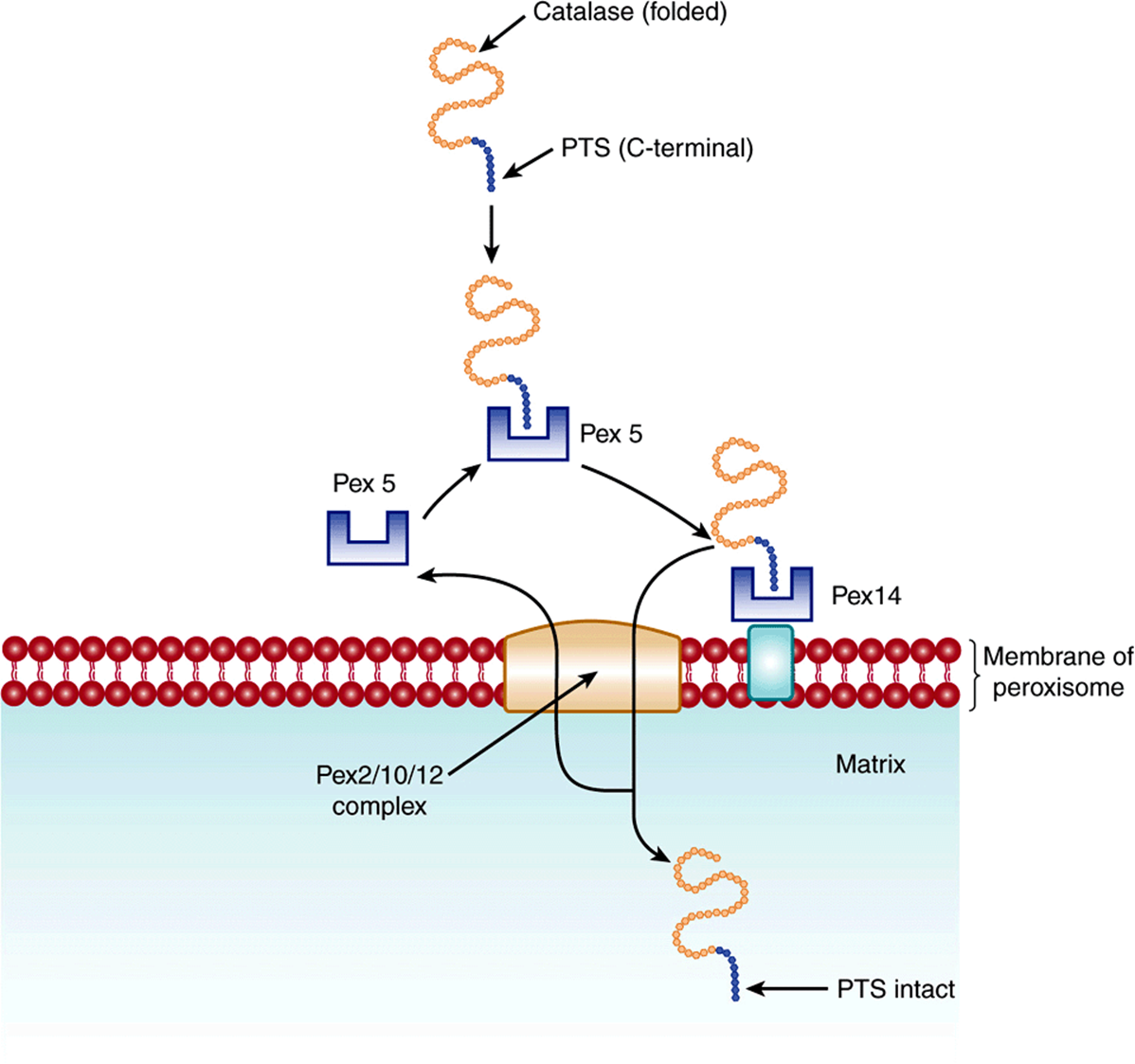
FIGURE 46–5 Schematic representation of the entry of a protein into the peroxisomal matrix. The protein to be imported into the matrix is synthesized on cytosolic polyribosomes, assumes its folded shape prior to import, and contains a C-terminal peroxisomal-targeting sequence (PTS). It interacts with cytosolic receptor protein Pex5, and the complex then interacts with a receptor on the peroxisomal membrane, Pex14. In turn, the protein-Pex 14 complex passes to the Pex 2/10/12 complex on the peroxisomal membrane and is translocated. Pex 5 is returned to the cytosol. The protein retains its PTS in the matrix. (Modified, with permission, from Lodish H, et al: Molecular Cell Biology, 6th ed. W.H. Freeman & Co., 2008.)
Most Cases of Zellweger Syndrome Are Due to Mutations in Genes Involved in the Biogenesis of Peroxisomes
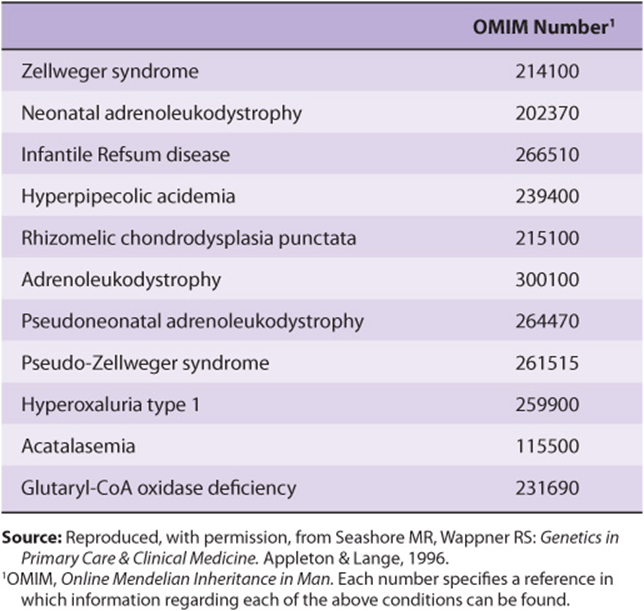
Interest in import of proteins into peroxisomes has been stimulated by studies on Zellweger syndrome. This condition is apparent at birth and is characterized by profound neurologic impairment, victims often dying within a year. The number of peroxisomes can vary from being almost normal to being virtually absent in some patients. Biochemical findings include an accumulation of very-long-chain fatty acids, abnormalities of the synthesis of bile acids, and a marked reduction of plasmalogens. The condition is believed to be due to mutations in genes encoding certain proteins—so-called peroxins— involved in various steps of peroxisome biogenesis (such as the import of proteins described above), or in genes encoding certain peroxisomal enzymes themselves. Two closely related conditions are neonatal adrenoleukodystrophy and infantile Refsum disease. Zellweger syndrome and these two conditions represent a spectrum of overlapping features, with Zellweger syndrome being the most severe (many proteins affected) and infantile Refsum disease the least severe (only one or a few proteins affected). Table 46-3 lists these and related conditions.
TABLE 46–3 Disorders Due to Peroxisomal Abnormalities
THE SIGNAL HYPOTHESIS EXPLAINS HOW POLYRIBOSOMES BIND TO THE ENDOPLASMIC RETICULUM
As indicated above, the rough ER branch is the second of the two branches involved in the synthesis and sorting of proteins. In this branch, proteins are synthesized on membrane-bound polyribosomes and are usually translocated into the lumen of the rough ER prior to further sorting (Figure 46–2). Certain membrane proteins, however, are transferred directly into the membrane of the ER without reaching its lumen.
The signal hypothesis was proposed by Blobel and Sabatini in 1971 partly to explain the distinction between free and membrane-bound polyribosomes. On the basis of certain experimental findings, they proposed that proteins synthesized on membrane-bound polyribosomes contained an N-terminal peptide extension (N-terminal signal peptide) which mediated their attachment to the membranes of the ER, and facilitated transfer into the ER lumen, On the other hand, proteins whose entire synthesis occurs on free polyribosomes would lack this signal peptide. An important aspect of the signal hypothesis was that it suggested—as turns out to be the case—that all ribosomes have the same structure and that the distinction between membrane-bound and free ribosomes depends solely on the former carrying proteins that have signal peptides. Because many membrane proteins are synthesized on membrane-bound polyribosomes, the signal hypothesis plays an important role in concepts of membrane assembly. Some characteristics of N-terminal signal peptides are summarized in Table 46-4.
TABLE 46–4 Some Properties of Signal Peptides Directing Proteins to the ER

There is much evidence to support the signal hypothesis, confirming that the N-terminal signal peptide is involved in the process of translocation across the ER membrane. For example, mutant proteins containing altered signal peptides in which hydrophobic amino acids are replaced by hydrophilic ones, are not inserted into the lumen of the ER. Non-membrane proteins (eg, α-globin) to which signal peptides have been attached by genetic engineering can be inserted into the lumen of the ER, or even secreted.
Many Details of the ER Translocation Process Have Been Revealed
Since the original formulation of the signal hypothesis, many scientists have contributed to revealing details of the overall process of translocation of nascent proteins across the ER membrane into its lumen. Table 46-5 lists some principal components of the overall process, and major steps in it are summarized in Figure 46–6.
TABLE 46–5 Principal Components Involved in ER Translocation
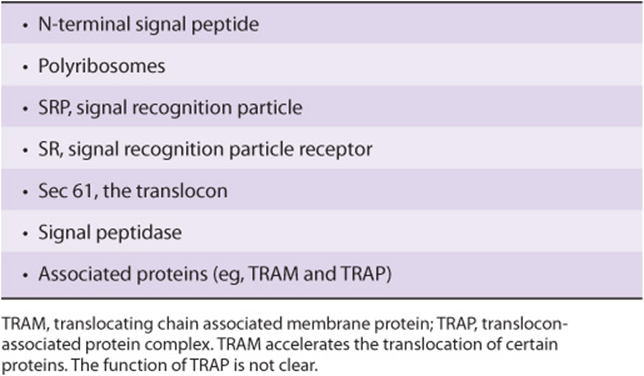
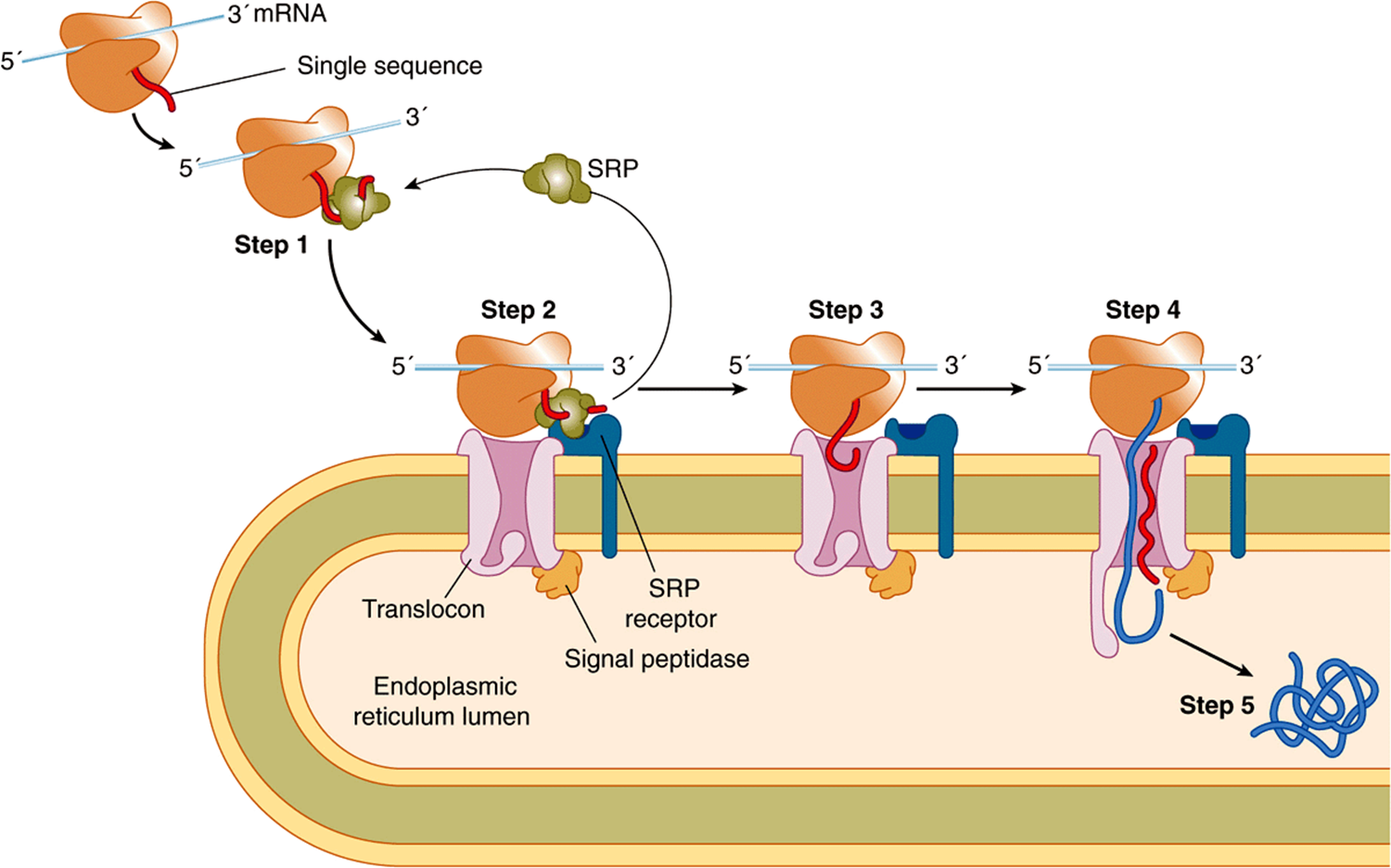
FIGURE 46–6 Cotranslational targeting of secretoryproteins to the ER. Step 1: As the signal sequence emerges from the ribosome, it is recognized and bound by the signal recognition particle (SRP). Step 2: The SRP escorts the complex to the ER membrane where it binds to the SRP receptor (SR). Step 3: The SRP is released, the ribosome binds to the translocon, and the signal sequence is inserted into the membrane channel. Step 4: The signal sequence opens the translocon. Translation resumes and the growing polypeptide chain is translocated across the membrane. Step 5: Cleavage of the signal sequence by signal peptidase relases the polypeptide into the lumen of the ER. Reproduced, with permission, from Cooper GM, Hausman RE: The Cell: A Molecular Approach. Sinauer Associates, Inc. 2009.
Step 1: The signal sequence emerges from the ribosome and binds to the SRP. This temporarily arrests further elongation of the polypeptide chain (elongation arrest) after some 70 amino acids have been polymerized.
Step 2: The SRP-ribosome-nascent protein complex travels to the ER membrane, where it binds to the SRP receptor (SRP-R). The SRP guides the complex to the SR, which prevents premature expulsion of the growing polypeptide into the cytosol.
Step 3: The SRP is released, translation resumes, the ribosome binds to the translocon (Sec 61 complex), and the signal peptide inserts into the channel in the translocon. As translation is still occurring, the entry of the signal peptide into the translocon and its further passage is termed cotranslational translocation.
Step 4: The signal peptide induces opening of the channel in the translocon by binding to certain hydrophobic residues in it, thus causing the plug (shown at the bottom on the translocon in Figure 46–6) to move. The growing polypeptide is then fully translocated across the membrane, driven by its ongoing synthesis.
Step 5: Cleavage of the signal peptide by signal peptidase occurs, and the fully translocated polypeptide/protein is released into the lumen of the ER. The signal peptide is presumably degraded by proteases. Ribosomes are released from the ER membrane and dissociate into their two types of subunits.
In yeast, many proteins are targeted to the ER after their translation is completed (posttranslational translocation) by a process that does not require the SRP. It does involve cytosolic chaperones (such as Hsp70) to keep the protein unfolded and also the luminal chaperone BiP, which may “pull” the growing polypeptide into the ER lumen. Some mammalian proteins also undergo this process.
Additional Comments on SRP, SRP Receptor, GTP, Sec61, and Glycosylation
The signal recognition particle (SRP) contains six proteins and has a 7S RNA associated with it that is closely related to the Alu family of highly repeated DNA sequences (Chapter 35). Both the RNA molecule and its proteins play various roles (such as binding other molecules) in its function.
The SRP receptor (SR) is an ER membrane protein composed of α and β subunits, the latter spanning the ER membrane.
SRP and both subunits of the SR can bind GTP. Both the SRP and SR must be in the GTP form to interact. When they bind, hydrolysis of GTP is stimulated, SRP is released, and the ribosome binds to the translocon allowing the signal peptide to enter it. The SRP and SR act as GTPase-acceleratng proteins (GAPs). When GTP is hydrolyzed to GDP, they dissociate. SRP and SR can be regarded as molecular matchmakers. SRP picks up the ribosome with its nascent chain and exposed signal peptide and the SR associates with the empty translocon pore, likely via its β subunit. The overall result of their interaction is to bring the ribosome to the translocon.
The translocon consists of three membrane proteins (the Sec61 complex) that form a protein-conducting channel in the ER membrane through which the newly synthesized protein may pass. As mentioned above, the channel appears to be open only when a signal peptide is present, preserving conductance across the ER membrane when it closes. The conductance of the channel has been measured experimentally. Closure of the channel when proteins are not being translocated prevents ions such as calcium and other molecules leaking through it, and causing cell dysfunction.
The insertion of the signal peptide into the conducting channel, while the other end of the parent protein is still attached to ribosomes, is termed cotranslational insertion. The process of elongation of the remaining portion of the protein being synthesized probably facilitates passage of the nascent protein across the lipid bilayer. It is important that proteins be kept in an unfolded state prior to entering the conducting channel—otherwise, they may not be able to gain access to the channel.
Secretory proteins and soluble proteins destined for organelles distal to the ER completely traverse the membrane bilayer and are discharged into the lumen of the ER. Many secretory proteins are N-glycosylated. N-Glycan chains, if present, are added by the enzyme oligosaccharide:protein transferase (Chapter 47) as these proteins traverse the inner part of the ER membrane—a process called cotranslational glycosylation. Subsequently, these glycoproteins are found in the lumen of the Golgi apparatus, where further changes in glycan chains occur (Figure 47–9) prior to intracellular distribution or secretion.
In contrast, proteins embedded in membranes of the ER as well as in other membranes along the secretory pathway only partially translocate across the ER membrane (see below). They are able to insert into the ER membrane by lateral transfer through the wall of the translocon.
There is evidence that the ER membrane is involved in retrograde transport of various molecules from the ER lumen to the cytosol. These molecules include unfolded or misfolded glycoproteins, glycopeptides, and oligosaccharides. At least some of these molecules are degraded in proteasomes (see below). The involvement of the translocon in retrotranslocation is not clear; one or more other channels may be involved. Whatever the case, there is two-way traffic across the ER membrane.
PROTEINS FOLLOW SEVERAL ROUTES TO BE INSERTED INTO OR ATTACHED TO THE MEMBRANES OF THE ENDOPLASMIC RETICULUM
The routes that proteins follow to be inserted into the membranes of the ER include the following.
Cotranslational Insertion
Figure 46–7 shows a variety of ways in which proteins are distributed in membranes. In particular, the amino termini of certain proteins (eg, the LDL receptor) can be seen to be on the extracytoplasmic face, whereas for other proteins (eg, the asialoglycoprotein receptor) the carboxyl termini are on this face. To explain these dispositions, one must consider the initial biosynthetic events at the ER membrane. The LDL receptor enters the ER membrane in a manner analogous to a secretory protein (Figure 46–6); it partly traverses the ER membrane, its signal peptide is cleaved, and its amino terminal protrudes into the lumen (see also Figure 46–13). However, it is retained in the membrane because it contains a highly hydrophobic segment, the halt- or stop-transfer signal (compare Figure 46–8). This sequence forms the single transmembrane segment of the protein and is its membrane-anchoring domain. The small patch of ER membrane in which the newly synthesized LDL receptor is located subsequently buds off as a component of a transport vesicle. As described below in the discussion of asymmetry of proteins and lipids in membrane assembly, the disposition of the receptor in the ER membrane is preserved in the vesicle (see Figure 46–13), which eventually fuses with the plasma membrane. In contrast, the asialoglycoprotein receptor lacks a cleavable N-terminal signal peptide, but possesses an internal insertion sequence, which inserts into the membrane but is not cleaved. This acts as an anchor, and its carboxyl terminus is extruded through the membrane. The more complex disposition of a transporter (eg, for glucose) can be explained by the fact that alternating transmembrane α-helices act as uncleaved insertion sequences and as halt-transfer signals, respectively. Each pair of helical segments is inserted as a hairpin. Sequences that determine the structure of a protein in a membrane are called topogenic sequences. As explained in the legend to Figure 46–7, the above three proteins are examples of type I, type II, and type IV transmembrane proteins, whereas cytochrome P450 is a member of type III.
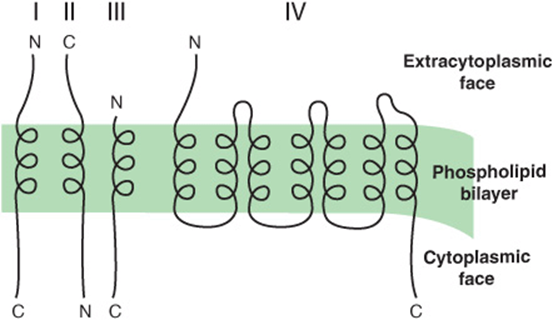
FIGURE 46–7 Variations in the way in which proteins are inserted into membranes. This schematic representation, which illustrates a number of possible orientations, shows the segments of the proteins within the membrane as α-helices and the other segments as lines. The orientations form initially in the ER membrane. Type I transmembrane proteins (eg, the LDL receptor and influenza hemagglutinin) cross the membrane once and have their amino termini on the exterior aspect of the membrane. Type II transmembrane proteins (eg, the asialoglycoprotein and transferrin receptors) also cross the membrane once, but have their C-termini on the exterior aspect. Type III transmembrane proteins (eg, cytochrome P450) have a disposition similar to type I proteins, but do not contain a cleavable signal peptide. Type IV transmembrane proteins (eg, G-protein-coupled receptors and glucose transporters) cross the membrane a number of times (7 times for the former and 12 times for glucose transporters); they are also called polytopic membrane proteins. (C, carboxyl terminal; N, amino terminal.) (Adapted, with permission, from Wickner WT, Lodish HF (1985), “Multiple mechanisms of protein insertion into and across membranes” Science 230:400. Reprinted with permission from AAAS.)
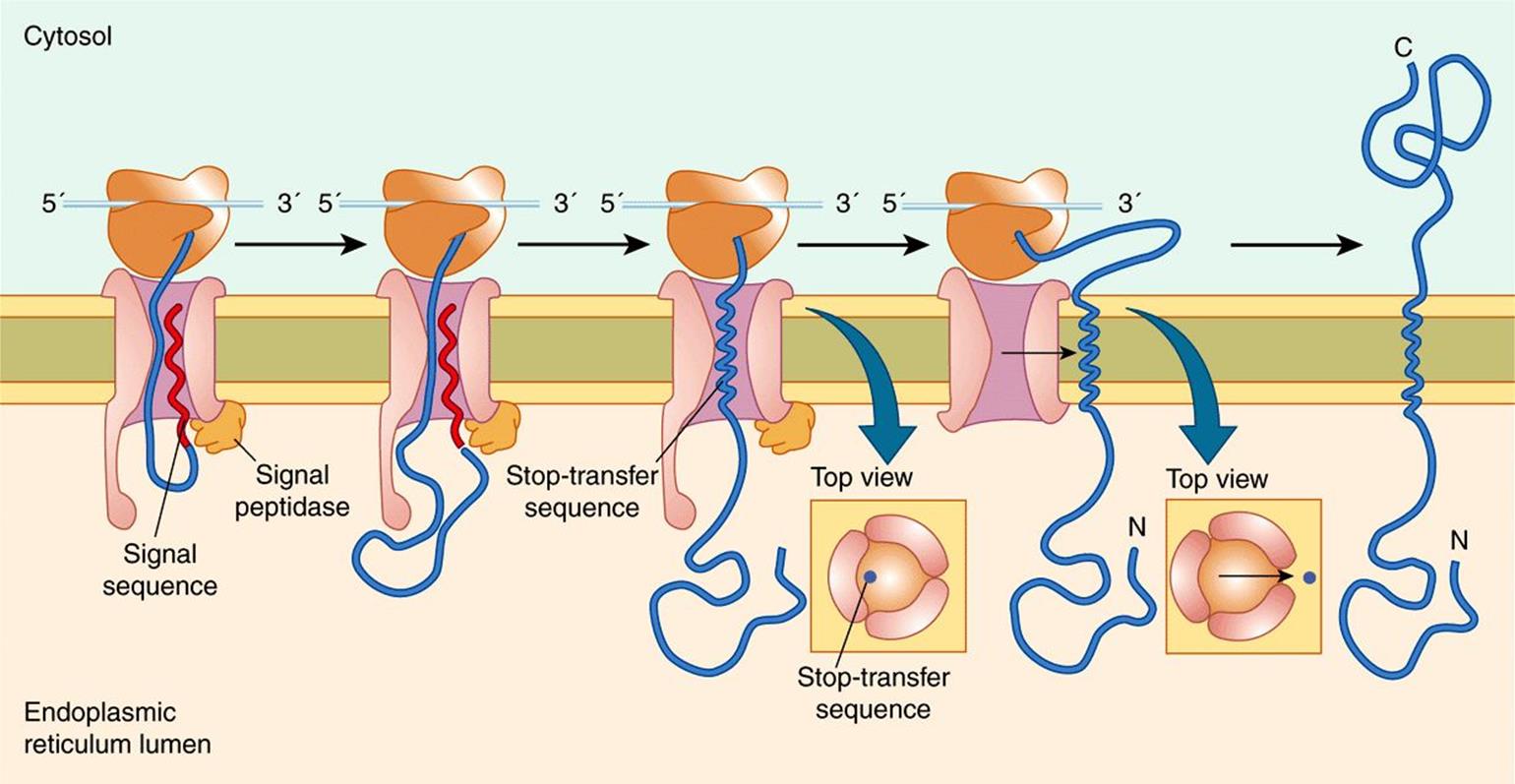
FIGURE 46–8 Insertion of a membrane protein with a cleavable signal sequence and a single stop-transfer sequence. The signal sequence is cleaved as the polypeptide chain crosses the membrane, so the amino terminus of the polypeptide chain is exposed in the ER lumen. However, translocation of the polypeptide chain across the membrane is halted when the translocon recognizes a transmembrane stop-transfer sequence. This closes the translocon and allows the protein to exit the channel laterally and become anchored in the ER membrane. Continued translation results in a membrane-spanning protein with its carboxy terminus on the cytosolic side. Reproduced with permission from Cooper GM Hausman RE: The Cell: A Molecular Approach. Sinauer Associates, Inc. 2009.
Synthesis on Free Polyribosomes & Posttranslational Attachment to the Endoplasmic Reticulum Membrane
An example is cytochrome b5, which appears to directly enter the ER membrane subsequent to translation, assisted by several chaperones.
Other Routes Include Retention in the GA with Retrieval to the ER and also Retrograde Transport from the GA
A number of proteins possess the amino acid sequence KDEL (Lys-Asp-Glu-Leu) at their carboxyl terminal (see Table 46-1). KDEL-containing proteins first travel to the GA in COPII transport vesicles (see below) and interact there with a specific KDEL receptor protein, which retains them transiently. They then return in COPI transport vesicles to the ER, where they dissociate from the receptor, and are thus retrieved. HDEL sequences (H = histidine) serve a similar purpose. The above processes result in net localization of certain soluble proteins to the ER lumen.
Certain other non-KDEL-containing proteins also pass to the Golgi and then return, by retrograde vesicular transport, to the ER to be inserted therein These include vesicle components that must be recycled, as well as certain ER membrane proteins. These proteins often possess a C-terminal signal located in the cytosol rich in basic residues.
The foregoing paragraphs demonstrate that a variety of routes are involved in assembly of the proteins of the ER membranes. A similar situation probably holds for other membranes (eg, the mitochondrial membranes and the plasma membrane). Precise targeting sequences have been identified in some instances (eg, KDEL sequences).
The topic of membrane biogenesis is discussed further later in this chapter.
CHAPERONES ARE PROTEINS THAT PREVENT FAULTY FOLDING & UNPRODUCTIVE INTERACTIONS OF OTHER PROTEINS
Molecular chaperones have been referred to previously in this Chapter. A number of important properties of these proteins are listed in Table 46-6, and the names of some of particular importance in the ER are listed in Table 46-7.Basically, they stabilize unfolded or partially folded intermediates, allowing them time to fold properly, and prevent inappropriate interactions, thus combating the formation of nonfunctional structures. Most chaperones exhibit ATPase activity and bind ADP and ATP. This activity is important for their effect on protein folding. The ADP-chaperone complex often has a high affinity for the unfolded protein, which, when bound, stimulates release of ADP with replacement by ATP. The ATP-chaperone complex, in turn, releases segments of the protein that have folded properly, and the cycle involving ADP and ATP binding is repeated until the protein is released.
TABLE 46–6 Some Properties of Chaperone Proteins

TABLE 46–7 Some Chaperones and Enzymes Involved in Folding That Are Located in the Rough Endoplasmic Reticulum

Chaperonins are the second major class of chaperones. They form complex barrel-like structures in which an unfolded protein is sequestered away from other proteins, giving it time and suitable conditions in which to fold properly. The structure of the bacterial chaperonin GroEL has been studied in detail. It is polymeric, has two ring-like structures, each composed of seven identical subunits, and again ATP is involved in its action.
Several examples of chaperones were introduced above when the sorting of mitochondrial proteins was discussed. The immunoglobulin heavy chain-binding protein (BiP) is located in the lumen of the ER. This protein promotes proper folding by preventing aggregation and will temporarily bind abnormally folded immunoglobulin heavy chains and many other proteins, preventing them from leaving the ER. Another important chaperone is calnexin, a calcium-binding protein located in the ER membrane. This protein binds a wide variety of proteins, including major histocompatibility complex (MHC) antigens and a variety of plasma proteins. As described in Chapter 47, calnexin binds the monoglucosylated species of glycoproteins that occur during processing of glycoproteins, retaining them in the ER until the glycoprotein has folded properly. Calreticulin, which is also a calcium-binding protein, has properties similar to those of calnexin; it is not membrane-bound. Chaperones are not the only proteins in the ER lumen that are concerned with proper folding of proteins. Two enzymes are present that play an active role in folding. Protein disulfide isomerase (PDI) promotes rapid formation and reshuffling of disulfide bonds until the correct set is achieved. Peptidyl prolyl isomerase (PPI) accelerates folding of proline-containing proteins by catalyzing the cis-trans isomerization of X-Pro bonds, where X is any amino acid residue.
Thus, the ER functions as a quality control compartment of the cell. Newly synthesized proteins attempt to fold with the assistance of chaperones and folding enzymes, and their folding status is monitored by the chaperones. Misfolded or incompletely folded proteins interact with chaperones, which retain them in the ER and prevent them from being exported to their final destinations. If such interactions continue for a prolonged period of time, the misfolded proteins are usually disposed of by endoplasmic reticulum-associated degradation (ERAD, see below). This avoids a harmful build-up of misfolded proteins. In a number of genetic diseases, such as cystic fibrosis (see Chapter 57), retention of misfolded proteins occurs in the ER, In some cases, the retained proteins still exhibit some functional activity. As discussed later in this Chapter, there is much current interest in finding drugs that will interact with such proteins and promote their correct folding and export out of the ER.
ACCUMULATION OF MISFOLDED PROTEINS IN THE ENDOPLASMIC RETICULUM CAN INDUCE THE UNFOLDED PROTEIN RESPONSE (UPR)
Maintenance of homeostasis in the ER is important for normal cell function. When the unique environment within the lumen of the ER is perturbed (eg, changes in ER Ca2+, alterations of redox status, exposure to various toxins or some viruses), this can lead to reduced protein folding capacity and the accumulation of misfolded proteins. The accumulation of misfolded proteins in the ER is referred to as ER stress. The cell has evolved a mechanism termed the UPR to sense the levels of misfolded proteins and initiate intracellular signaling mechanisms to compensate for the stress conditions and restore ER homeostasis. The UPR is initiated by ER stress sensors, which are transmembrane proteins embedded in the ER membrane. Activation of these stress sensors causes three principal effects: (i) transient inhibition of translation to reduce the amount of newly synthesized proteins, (ii) induction of a transcriptional response that leads to increased expression of ER chaperones and to (iii) increased synthesis of proteins involved in degradation of misfolded ER proteins (discussed below). Therefore, the UPR increases the ER folding capacity and prevents a buildup of unproductive and potentially toxic protein products, in addition to other responses to restore cellular homeostasis. However, if impairment of folding persists, cell death pathways (apoptosis) are activated. A more complete understanding of the UPR is likely to provide new approaches to treating diseases in which ER stress and defective protein folding occur (see Table 46-8).
TABLE 46–8 Some Conformational Diseases That Are Caused by Abnormalities in Intracellular Transport of Specific Proteins and Enzymes Due to Mutations1
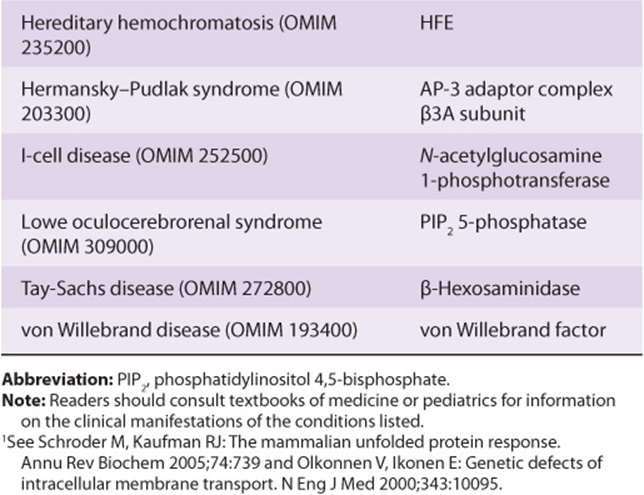
MISFOLDED PROTEINS UNDERGO ENDOPLASMIC RETICULUM-ASSOCIATED DEGRADATION
As shown in Table 46-8, misfolded proteins occur in many genetic diseases. Proteins that misfold in the ER are selectively transported back across the ER (retrotranslocation or dislocation) to enter proteasomes present in the cytosol. The precise route by which the misfolded proteins pass back across the ER membrane is still under investigation. Two proteins have been implicated: the Sec61 complex described earlier and Derlin 1. The energy for translocation appears to be at least partly supplied by p97, an AAA-ATPase (one of a family of ATPases A ssociated with various cellular Activities). Chaperones present in the lumen of the ER (eg, BiP) and in the cytosol help target misfolded proteins to proteasomes. Prior to entering proteasomes, most proteins are ubiquitinated (see the next paragraph) and are escorted to proteasomes by polyubiquitin-binding proteins. Ubiquitin ligases are present in the ER membrane. The above process is referred to as ERAD and is outlined briefly in Figure 46–9.
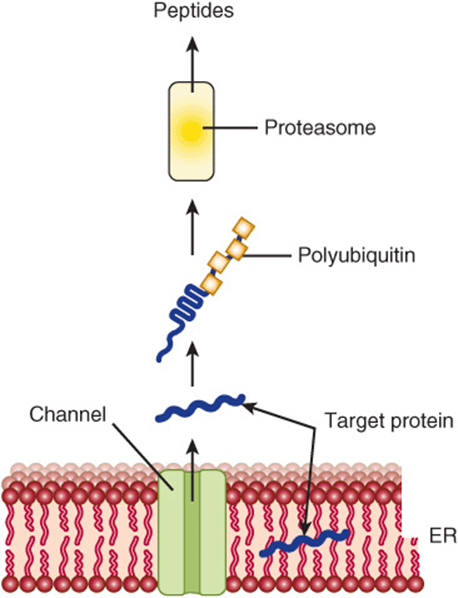
FIGURE 46–9 Simplified schematic diagram of the events in ERAD. A target protein which is misfolded undergoes retrograde transport through the ER membrane into the cytosol, where it is subjected to polyubiquitination. Following polyubiquitination, it enters a proteasome, inside which it is degraded to small peptides that exit and may have several fates. Liberated ubiquitin molecules are recycled. Two proteins have been implicated in the retrograde transport through the ER; these are Sec 61 (also involved in transfer of newly synthesized proteins into the lumen of the ER) and another protein named Derlin 1.
UBIQUITIN IS A KEY MOLECULE IN PROTEIN DEGRADATION
There are two major pathways of protein degradation in eukaryotes. One involves lysosomal proteases and does not require ATP. The other pathway involves ubiquitin and is ATP-dependent. It plays the major role in the degradation of proteins, and is particularly associated with disposal of misfolded proteins and regulatory enzymes that have short half-lives. Research on ubiquitin has expanded rapidly, and it is known to be involved in cell-cycle regulation (degradation of cyclins), DNA repair, activation of NFKB (see Chapter 50), muscle wasting, viral infections, and many other important physiologic and pathologic processes. Ubiquitin is a small (76 amino acids), highly conserved protein that plays a key role in marking various proteins for subsequent degradation in proteasomes. The mechanism of attachment of ubiquitin to a target protein (eg, a misfolded form of CFTR, the protein involved in the causation of cystic fibrosis; see Chapters 40 and 57) is shown in Figure 46–10 and involves three enzymes: an activating enzyme, a conjugating enzyme, and a ligase. There are a number of types of conjugating enzymes, and, surprisingly, some hundreds of different ligases. It is the latter enzyme that confers substrate specificity. Once the molecule of ubiq-uitin is attached to the protein, a number of others are also attached, resulting in a polyubiquitinated target protein. It has been estimated that a minimum of four ubiquitin molecules must be attached to commit a target molecule to degradation in a proteasome. Ubiquitin can be cleaved from a target protein by deubiquitinating enzymes and the liberated ubiquitin can be reused.
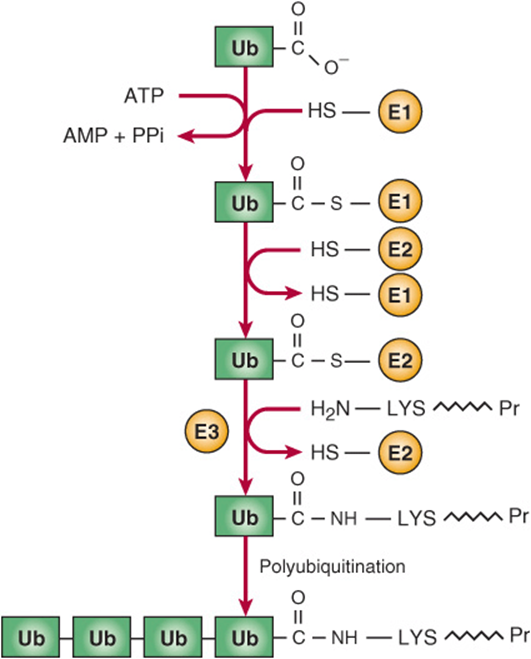
FIGURE 46–10 Sequence of reactions in addition of ubiquitin to a target protein. In the reaction catalyzed by E1, the C-terminal COO– group of ubiquitin is linked in a thioester bond to an SH group of E1. In the reaction catalyzed by E2, the activated ubiquitin is transferred to an SH group of E2. In the reaction catalyzed by E3, ubiquitin is transferred from E2 to an ε-amino group on a lysine of the target protein. Additional rounds of ubiquitination then build up the polyubiquitin chain. (Ub, ubiquitin; E1, activating enzyme; E2, conjugating enzyme; E3, ligase; LYS ![]() Pr, target protein.)
Pr, target protein.)
Ubiquitinated Proteins Are Degraded in Proteasomes
Polyubiquitinated target proteins enter proteasomes located in the cytosol. The proteasome is a relatively large cylindrical structure and is composed of some 50 subunits. The proteasome has a hollow core, and one or two capsthat play a regulatory role. Target proteins are unfolded by ATPases present in the proteasome caps. Proteasomes can hydrolyze a very wide variety of peptide bonds. Target proteins pass into the core to be degraded to small peptides, which then exit the proteasome (see Figure 46–9) to be further degraded by cytosolic peptidases. Both normally and abnormally folded proteins are substrates for the proteasome. Liberated ubiquitin molecules are recycled. The proteasome plays an important role in presenting small peptides produced by degradation of various viruses and other molecules to major histocompatibility class I molecules, a key step in antigen presentation to T lymphocytes.
TRANSPORT VESICLES ARE KEY PLAYERS IN INTRACELLULAR PROTEIN TRAFFIC
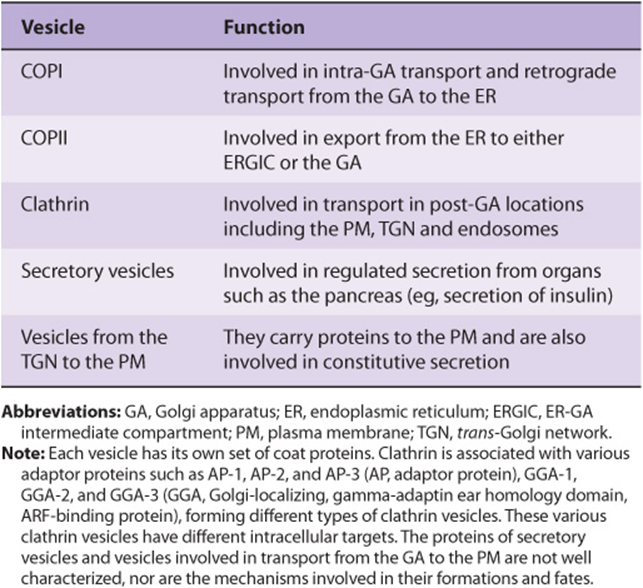
Proteins that are synthesized on membrane-bound polyribosomes and are destined for the GA or PM reach these sites inside transport vesicles. Those vesicles involved in anterograde transport (COPII) from the ER to the GA and in retrograde transport (COPI) from the GA to the ER are clathrin-free. Transport and secretory vesicles carrying cargo from the GA to the PM are also clathrin-free. The vesicles involved in endocytosis (see discussions of the LDL receptor in Chapters 25 and 26) are coated with clathrin, as are certain vesicles carrying cargo to lysosomes. For the sake of clarity, the non-clathrin-coated vesicles are referred to in this text as transport vesicles. Table 46-9summarizes the types and functions of the major vesicles identified to date.
TABLE 46–9 Some Types of Vesicles and Their Functions
Model of Transport Vesicles Involves SNAREs & Other Factors
Vesicles lie at the heart of intracellular transport of many proteins. Significant progress has been made in understanding the events involved in vesicle formation and transport. This has transpired because of the use of a number of approaches. In particular, the use by Schekman and colleagues of genetic approaches for studying vesicles in yeast and the development by Rothman and colleagues of cell-free systems to study vesicle formation have been crucial. For instance, it is possible to observe, by electron microscopy, budding of vesicles from Golgi preparations incubated with cytosol, ATP and GTP-γ. The overall mechanism is complex, with its own nomenclature (Table 46-10), and involves a variety of cytosolic and membrane proteins, GTP, ATP, and accessory factors. Budding, tethering, docking, and membrane fusion are key steps in the life cycles of vesicles with Sar, ARF, and the Rab GTPases (see below) acting as molecular switches.
TABLE 46–10 Some Factors Involved in the Formation of Non-clathrin-coated Vesicles and Their Transport

There are common general steps in transport vesicle formation, vesicle targeting and fusion with a target membrane, irrespective of the membrane the vesicle forms from or its intracellular destination. The nature of the coat proteins, GTPases and targeting factors differ depending on where the vesicle forms from and its eventual destination. Transport from the ER to the Golgi is the best studied example and will be used to illustrate these steps. Anterograde vesicular transport from the ER to the Golgi involves COPII vesicles and the process can be considered to occur in eight steps (Figure 46–11). The basic concept is that each transport vesicle is loaded with specific cargo and also one or more v-SNARE proteins that direct targeting. Each target membrane bears one or more complementary t-SNARE proteins with which the former interact, mediating SNARE protein-dependent vesicle-membrane fusion. In addition, Rab proteins also help direct the vesicles to specific membranes and are involved in tethering, prior to vesicle docking at a target membrane.
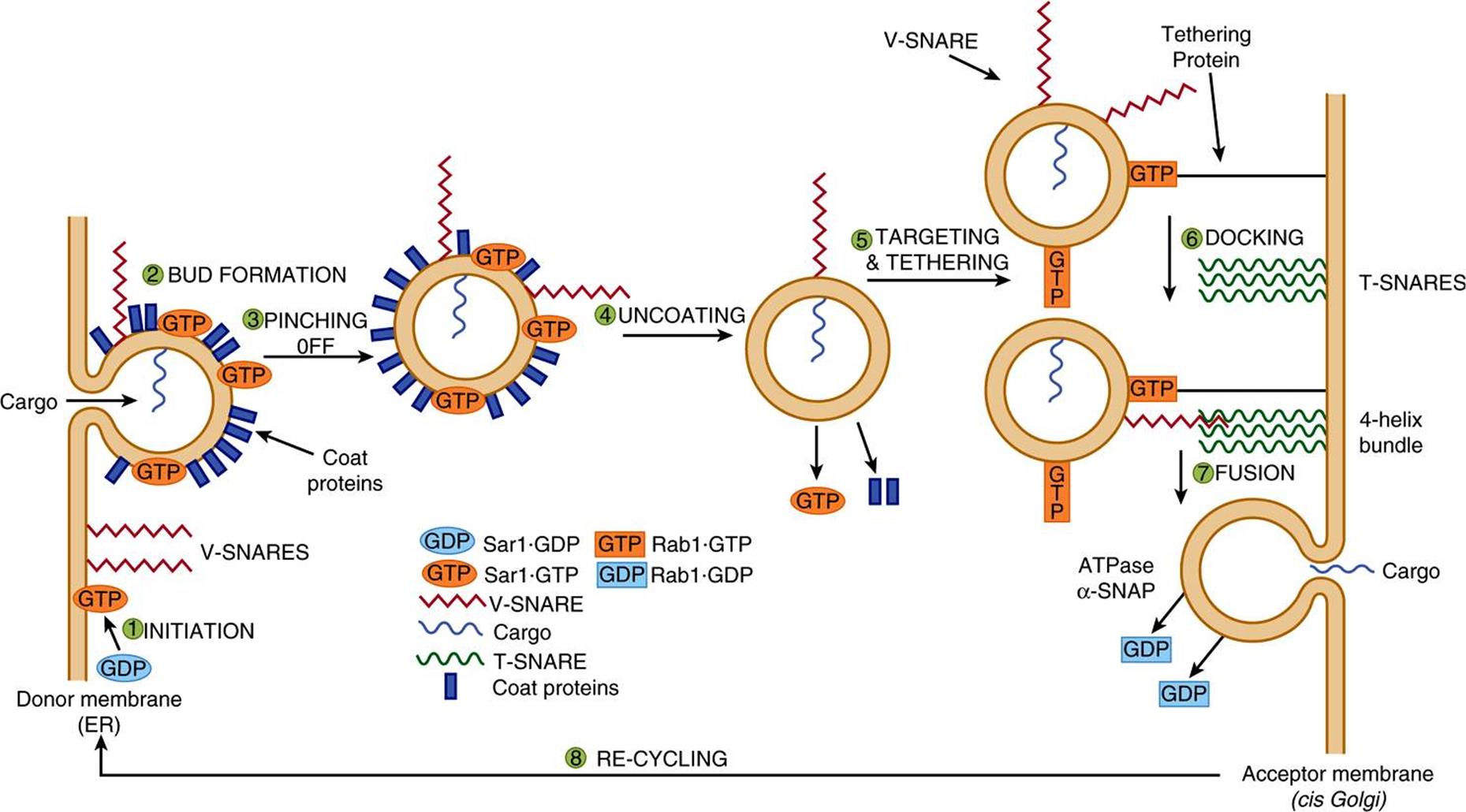
FIGURE 46–11 Model of the steps in a round of anterograde transport involving COPII vesicles. The cycle starts in the bottom left-hand side of the figure, where a molecule of Sar1·GDP is represented as a blue oval labelled GDP. The steps in the cycle are described in the text. The various components are briefly described in Table 46-8. The roles of Rab and Rab effector proteins (see text) in the overall process are not dealt with in this figure. (Adapted, with permission, from Rothman JE: Mechanisms of intracellular protein transport. Nature 1994;372:55.)
Step 1: Budding is initiated when Sar1 is activated by binding GTP, which is exchanged for GDP via the action of Sec12. This causes a conformational change in Sar1-GTP, embedding it in the ER membrane to form a focal point for vesicle assembly.
Step 2: Various coat proteins bind to Sar1-GTP. In turn, membrane cargo proteins bind to the coat proteins and soluble cargo proteins inside vesicles bind to receptor regions of the former. Additional coat proteins are assembled to complete bud formation. Coat proteins promote budding, contribute to the curvature of buds and also help sort proteins.
Step 3: The bud pinches off, completing formation of the coated vesicle. The curvature of the ER membrane and protein-protein and protein-lipid interactions in the bud facilitate pinching off from ER exit sites.
Step 4: Coat disassembly (involving dissociation of Sar1 and the shell of coat proteins) follows hydrolysis of bound GTP to GDP by Sar1, promoted by a specific coat protein. Sar1 thus plays key roles in both assembly and dissociation of the coat proteins. Uncoating is necessary for fusion to occur.
Step 5: Vesicle targeting is achieved by attachment of Rab molecules to vesicles. Rab-GDP molecules in the cytosol are converted to Rab-GTP molecules by a specific GEF and these attach to the vesicles. The Rab-GTP molecules subsequently interact with Rab effector proteins on membranes to tether the vesicle to the membranes.
Step 6: v-SNAREs pair with cognate t-SNAREs in the target membrane to dock the vesicles and initiate fusion. Generally one v-SNARE in the vesicle pairs with three t-SNAREs on the acceptor membrane to form a tight four-helix bundle.
Step 7: Fusion of the vesicle with the acceptor membrane occurs once the v- and t-SNARES are closely aligned. After vesicle fusion and release of contents occurs, GTP is hydrolyzed to GDP, and the Rab-GDP molecules are released into the cytosol. When a SNARE on one membrane interacts with a SNARE on another membrane, linking the two membranes, this is referred to as a trans-SNARE complex or a SNARE pin. Interactions of SNARES on the same membrane form a cis-SNARE complex. In order to dissociate the four-helix bundle between the v- and t-SNARES so that they can be reused, two additional proteins are required. These are an ATPase (NSF) and α-SNAP. NSF hydrolyzes ATP and the energy released dissociates the four-helix bundle making the SNARE proteins available for another round of membrane fusion.
Step 8: Certain components, such as the Rab and SNARE proteins, are recycled for subsequent rounds of vesicle fusion.
During the above cycle, SNARES, tethering proteins, Rab and other proteins all collaborate to deliver a vesicle and its contents to the appropriate site.
COPI, COPII, and Clathrin-Coated Vesicles Have Been Most Studied
The following points clarify and expand on the previous section.
1. As indicated in Table 46-9, there are a number of different types of vesicles. Other types of vesicles may remain to be discovered. Here we focus mainly on COPII, COPI and clathrin-coated vesicles. Each of these types has a different complement of proteins in its coat. The details of assembly for COPI and clathrin-coated vesicles are somewhat different from those described above. For example, Sar1 is the protein involved in step 1 of formation of COPII vesicles, whereas ARF is involved in the formation of COPI and clathrin-coated vesicles. However, the principles concerning assembly of these different types are generally similar.
2. Regarding selection of cargo molecules by vesicles, this appears to be primarily a function of the coat proteins of vesicles. Cargo molecules via their sorting signals may interact with coat proteins either directly or via intermediary proteins that attach to coat proteins, and they then become enclosed in their appropriate vesicles. A number of signal sequences on cargo molecules have been identified (see Table 46-1). For example KDEL sequences direct certain ER-resident proteins in retrograde flow to the ER in COPI vesicles. Di-acidic sequences (eg, Asp-X-Glu, X = any amino acid) and short hydrophobic sequences on membrane proteins are involved in interactions with coat proteins of COPII vesicles.
Proteins in the apical or basolateral areas of the plasma membranes of polarized epithelial cells can be transported to these sites in transport vesicles budding from the TGN. Different Rab proteins likely direct some vesicles to apical regions and others to basolateral regions. In certain cells, proteins are first directed to the basolateral membrane, then endocytosed and transported across the cell by transcytosis to the apical region. Yet another mechanism for sorting proteins to the apical region (or in some cases to the basolateral region) involves the glycosylphosphatidylinositol (GPI) anchor described in Chapter 47, This structure is also often present in lipid rafts (see Chapter 40).
Not all cargo molecules may have a sorting signal. Some highly abundant secretory proteins travel to various cellular destinations in transport vesicles by bulk flow; that is, they enter into transport vesicles at the same concentration that they occur in the organelle. The precise extent of bulk flow is not clearly known, although it appears that most proteins are actively sorted (concentrated) into transport vesicles and bulk flow is used by only a select group of cargo proteins.
3. Once proteins in the secretory pathway reach the cis-Golgi from the ER in vesicles, they can travel through the GA to the trans-Golgi in vesicles, or by a process called cisternal maturation, or perhaps in some cases diffusionvia intracisternal connections that have been observed in some cell types. A former view was that the GA is essentially a static organelle, allowing vesicular flow from one static cisterna to the next. There is now, however, evidence to support the view that the cisternae move and transform into one another (ie, cisternal maturation). In this model, vesicular elements from the ER fuse with one another to help form the cis-Golgi, which in turn can move forward to become the medial Golgi, etc. COPI vesicles return Golgi enzymes (eg, glycosyltransferases) back from distal cisternae of the GA to more proximal (eg, cis) cisternae.
4. Vesicles move through cells along microtubules or along actin filaments.
5. The fungal metabolite brefeldin A prevents GTP from binding to ARF, and thus inhibits formation of COPI vesicles. In its presence, the Golgi apparatus appears to collapse into the ER. It may do this by inhibiting the guanine nucleotide exchanger involved in formation of COPI vesicles. Brefeldin A has thus proven to be a useful tool for examining some aspects of Golgi structure and function.
6. GTP-γ-S (a nonhydrolyzable analog of GTP often used in investigations of the role of GTP in biochemical processes) blocks disassembly of the coat from coated vesicles, leading to a build-up of coated vesicles, facilitating their study.
7. As mentioned above, a family of Ras-like proteins, called the Rab protein family, is required in several steps of intracellular protein transport and also in regulated secretion and endocytosis. (Ras proteins are involved in cell signaling via receptor tyrosine kinases). Like Ras, Rab proteins are small monomeric GTPases that attach to the cytosolic faces of membranes (via geranylgeranyl lipid anchors). They attach in the GTP-bound state to the budding vesicle and are also present on acceptor membranes. Rab proteins interact with Rab effector proteins that have various roles, such as involvement in tethering and in membrane fusion.
8. The fusion of synaptic vesicles with the plasma membrane of neurons involves a series of events similar to that described above. For example, one v-SNARE is designated synaptobrevin and two t-SNAREs are designated syntaxin and SNAP 25 (synaptosome-associated protein of 25 kDa). Botulinum B toxin is one of the most lethal toxins known and the most serious cause of food poisoning. One component of this toxin is a protease that appears to cleave only synaptobrevin, thus inhibiting release of acetylcholine at the neuromuscular junction and possibly proving fatal, depending on the dose taken.
9. Although the above model refers to non-clathrin-coated vesicles, it appears likely that many of the events described above apply, at least in principle, to clathrin-coated vesicles.
10. Some proteins are further subjected to further processing by proteolysis while inside either transport or secretory vesicles. For example, albumin is synthesized by hepatocytes as preproalbumin (see Chapter 50). Its signal peptide is removed, converting it to proalbumin. In turn, proalbumin, while inside transport vesicles, is converted to albumin by action of furin (Figure 46–12). This enzyme cleaves a hexapeptide from proalbumin immediately C-terminal to a dibasic amino acid site (ArgArg). The resulting mature albumin is secreted into the plasma. Hormones such as insulin (see Chapter 41) are subjected to similar proteolytic cleavages while inside secretory vesicles.
![]()
FIGURE 46–12 Cleavage of preproalbumin to proalbumin and of the latter to albumin. Furin cleaves proalbumin at the C-terminal end of a basic dipeptide (ArgArg).
THE ASSEMBLY OF MEMBRANES IS COMPLEX
There are many cellular membranes, each with its own specific features. No satisfactory scheme describing the assembly of any one of these membranes is available. How various proteins are initially inserted into the membrane of the ER has been discussed above. The transport of proteins, including membrane proteins, to various parts of the cell inside vesicles has also been described. Some general points about membrane assembly remain to be addressed.
Asymmetry of Both Proteins & Lipids Is Maintained During Membrane Assembly
Vesicles formed from membranes of the ER and Golgi apparatus, either naturally or pinched off by homogenization, exhibit transverse asymmetries of both lipid and protein. These asymmetries are maintained during fusion of transport vesicles with the plasma membrane. The inside of the vesicles after fusion becomes the outside of the plasma membrane, and the cytoplasmic side of the vesicles remains the cytoplasmic side of the membrane (Figure 46–13). Since the transverse asymmetry of the membranes already exists in the vesicles of the ER well before they are fused to the plasma membrane, a major problem of membrane assembly becomes understanding how the integral proteins are inserted into the lipid bilayer of the ER. This problem was addressed earlier in this chapter.
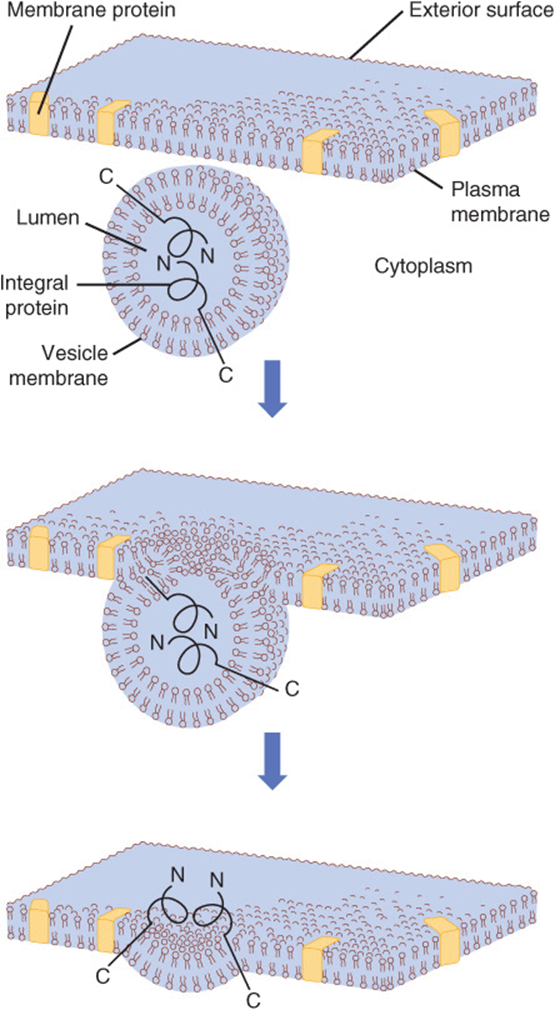
FIGURE 46–13 Fusion of a vesicle with the plasma membrane preserves the orientation of any integral proteins embedded in the vesicle bilayer. Initially, the amino terminal of the protein faces the lumen, or inner cavity, of such a vesicle. After fusion, the amino terminal is on the exterior surface of the plasma membrane. That the orientation of the protein has not been reversed can be perceived by noting that the other end of the molecule, the carboxyl terminal, is always immersed in the cytoplasm. The lumen of a vesicle and the outside of the cell are topologically equivalent. (Redrawn and modified, with permission, from Lodish HF, Rothman JE: The assembly of cell membranes. Sci Am [Jan] 1979;240:43.)
Phospholipids are the major class of lipid in membranes. The enzymes responsible for the synthesis of phospholipids reside in the cytoplasmic surface of the cisternae of the ER. As phospholipids are synthesized at that site, they probably self-assemble into thermodynamically stable bimolecular layers, thereby expanding the membrane and perhaps promoting the detachment of so-called lipid vesicles from it. It has been proposed that these vesicles travel to other sites, donating their lipids to other membranes; however, little is known about this matter. As indicated above, cytosolic proteins that take up phospholipids from one membrane and release them to another (ie, phospholipid exchange proteins) have been demonstrated; they probably play a role in contributing to the specific lipid composition of various membranes.
It should be noted that the lipid compositions of the ER, Golgi, and plasma membrane differ, the latter two membranes containing higher amounts of cholesterol, sphingomyelin, and glycosphingolipids, and less phosphoglycerides than does the ER. Sphingolipids pack more densely in membranes than do phosphoglycerides. These differences affect the structures and functions of membranes. For example, the thickness of the bilayer of the GA and PM is greater than that of the ER, which affects which particular transmembrane proteins are found in these organelles. Also, lipid rafts (see Chapter 40) are believed to be formed in the GA.
Lipids & Proteins Undergo Turnover at Different Rates in Different Membranes
It has been shown that the half-lives of the lipids of the ER membranes of rat liver are generally shorter than those of its proteins, so that the turnover rates of lipids and proteins are independent. Indeed, different lipids have been found to have different half-lives. Furthermore, the half-lives of the proteins of these membranes vary quite widely, some exhibiting short (hours) and others long (days) half-lives. Thus, individual lipids and proteins of the ER membranes appear to be inserted into it relatively independently; this is the case for many other membranes.
The biogenesis of membranes is thus a complex process about which much remains to be learned. One indication of the complexity involved is to consider the number of posttranslational modifications that membrane proteins may be subjected to prior to attaining their mature state. These include disulfide formation, proteolysis, assembly into multimers, glycosylation, addition of a glycophosphatidylinositol (GPI) anchor, sulfation on tyrosine or carbohydrate moieties, phosphorylation, acylation, and prenylation—a list that is not complete. Nevertheless, significant progress has been made; Table 46-11 summarizes some of the major features of membrane assembly that have emerged to date.
TABLE 46–11 Some Major Features of Membrane Assembly

Various Disorders Result from Mutations in Genes Encoding Proteins Involved in Intracellular Transport
Some disorders reflecting abnormal peroxisomal function and abnormalities of protein synthesis in the ER and of the synthesis of lysosomal proteins have been listed earlier in this chapter (see Tables 46-3 and 46-8, respectively). Many other mutations affecting folding of proteins and their intracellular transport to various organelles have been reported, but are not discussed here [eg, Alzheimer disease (see Chapter 57)] and Huntington disease. The elucidation of the causes of these various conformational disorders has contributed significantly to our understanding of molecular pathology. The term “diseases of proteostasis deficiency” has also been applied to diseases due to misfolding of proteins. Proteostasis is a composite word derived from protein homeostasis. Normal proteostasis is due to a balance of many factors, such as synthesis, folding, trafficking, aggregation, and normal degradation. If any one of these is disturbed (eg, by mutation, aging, cell stress or injury), a variety of disorders can occur, depending on the particular proteins involved.
Apart from the possibility of gene therapy, it is hoped that attempts to restore at least a degree of normal folding to misfolded proteins by administration to affected individuals of small molecules that interact specifically with such proteins will be of therapeutic benefit. This is a very active area of research.
SUMMARY
![]() Many proteins are targeted to their destinations by signal sequences. A major sorting decision is made when proteins are partitioned between cytosolic and membrane-bound polyribosomes by virtue of the absence or presence of an N-terminal signal peptide.
Many proteins are targeted to their destinations by signal sequences. A major sorting decision is made when proteins are partitioned between cytosolic and membrane-bound polyribosomes by virtue of the absence or presence of an N-terminal signal peptide.
![]() Pathways of protein import into mitochondria, nuclei, peroxisomes, and the endoplasmic reticulum are described.
Pathways of protein import into mitochondria, nuclei, peroxisomes, and the endoplasmic reticulum are described.
![]() Numerous proteins synthesized on membrane-bound polyribosomes proceed to the Golgi apparatus and the plasma membrane in transport vesicles.
Numerous proteins synthesized on membrane-bound polyribosomes proceed to the Golgi apparatus and the plasma membrane in transport vesicles.
![]() Many glycosylation reactions occur in compartments of the Golgi, and proteins are further sorted in the trans-Golgi network.
Many glycosylation reactions occur in compartments of the Golgi, and proteins are further sorted in the trans-Golgi network.
![]() The role of chaperone proteins in the folding of proteins is presented and the UPR is described.
The role of chaperone proteins in the folding of proteins is presented and the UPR is described.
![]() ERAD is briefly described and the key role of ubiquitin in protein degradation is shown.
ERAD is briefly described and the key role of ubiquitin in protein degradation is shown.
![]() A model describing budding and attachment of transport vesicles to a target membrane is summarized.
A model describing budding and attachment of transport vesicles to a target membrane is summarized.
![]() Certain proteins (eg, precursors of albumin and insulin) are subjected to proteolysis while inside transport vesicles, producing the mature proteins.
Certain proteins (eg, precursors of albumin and insulin) are subjected to proteolysis while inside transport vesicles, producing the mature proteins.
![]() Small GTPases (eg, Ran, Rab) and GEFs play key roles in many aspects of intracellular trafficking.
Small GTPases (eg, Ran, Rab) and GEFs play key roles in many aspects of intracellular trafficking.
![]() The complex process of membrane assembly is discussed briefly. Asymmetry of both lipids and proteins is maintained during membrane assembly.
The complex process of membrane assembly is discussed briefly. Asymmetry of both lipids and proteins is maintained during membrane assembly.
![]() Many disorders have been shown to be due to mutations in genes or to other factors that affect the folding of various proteins. These conditions have been referred to as conformational diseases, or alternatively as diseases of proteostatic deficiency. Apart from gene therapy, the development of small molecules that interact with misfolded proteins and help restore at least some of their function is an important area of research.
Many disorders have been shown to be due to mutations in genes or to other factors that affect the folding of various proteins. These conditions have been referred to as conformational diseases, or alternatively as diseases of proteostatic deficiency. Apart from gene therapy, the development of small molecules that interact with misfolded proteins and help restore at least some of their function is an important area of research.
REFERENCES
Alberts B, Johnson A, Lewis J, et al: Molecular Biology of the Cell, 5th ed. Garland Science, 2008. (An excellent textbook of cell biology, with comprehensive coverage of trafficking and sorting).
Alder NN, Johnson AE: Cotranslational membrane protein biogenesis at the endoplasmic reticulum. J Biol Chem 2004;279:22787.
Bonifacino JS, Glick BS: The mechanisms of vesicle budding and fusion. Cell 2004;116:153.
Cooper GM, Hausman RE: The Cell: A Molecular Approach. Sinauer Associates, Inc. 2009. (An excellent textbook of cell biology, with comprehensive coverage of trafficking and sorting).
Lai E, Teodoro T, Volchuk A: Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology 2007;22:193.
Lodish H, Berk A, Krieger M, et al: Molecular Cell Biology, 6th ed. WH Freeman & Co., 2008. (An excellent textbook of cell biology, with comprehensive coverage of trafficking and sorting).
Neupert W, Herrmann JM: Translocation of proteins into mitiochondria. Annu Rev Biochem 2007;76:723.
Platta HW, Erdmann R: The peroxisomal protein import machinery. FEBS Lett 2007;581;2811.
Pollard TD, Earnshaw WC: Cell Biology, 2nd ed. WB Saunders, 2008. (An excellent textbook of cell biology, with comprehensive coverage of trafficking and sorting).
Powers ET, Morimoto RI, Dillin A, et al: Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 2009;78:959.
Romisch K: Endoplasmic-reticulum-associated degradation. Annu Rev Cell Dev Biol 2005;21:435.
Stewart M: Molecular mechanisms of the nuclear protein import cycle. Nature Rev Mol Cell Biol 2007;8:195.