CHEMICAL BIOLOGY
Biosynthesis of Nonribosomal Peptides
Georg Schoenafinger, Philipps-University of Marburg, FB Chemie/Biochemie, Marburg, Germany
Mohamed A Marahiel, Philipps -University of Marburg, FB Chemie/Biochemie, Marburg, Germany
doi: 10.1002/9780470048672.wecb398
Many microorganisms have evolved an unusual way of producing secondary peptide metabolites. Large multidomain enzymatic machineries, the so-called nonribosomal peptide synthetases (NRPSs), are responsible for the production of this structurally diverse class of peptides with various functions, such as cytostatic, immunosuppressive, antibacterial, or antitumor properties. These secondary metabolites differ from peptides of ribosomal origin in several ways. Their length is limited to a mere 20 building blocks, roughly, and mostly a circular or branched cyclic connectivity is found. Furthermore, aside from the proteinogenic amino acids, a larger variety of chemical groups is found in these bioactive compounds: D-configurated amino acids, fatty acids, methylated, oxidized, halogenated, and glycosylated building blocks. These functional and structural features are known to be important for bioactivity, and often natural defense mechanisms are thus evaded. In this article, we describe the enzymatic machineries of NRPSs, the chemical reactions catalyzed by their subunits, and the potential of redesigning or using these machineries to give rise to new nonribosomal peptide antibiotics.
Nonribosomal peptide synthetases (NRPSs) compose a unique class of multidomain enzymes capable of producing peptides (1-4). In contrast to the ribosomal machinery where the mRNA template is translated, the order of catalytically active entities within these synthetases intrinsically determines the sequence of building blocks in the peptide product (Fig. 1). As a consequence, generally speaking, each NRPS can only produce one defined peptide product. This is chemically implemented by the fact that all substrates and reaction intermediates are spatially fixed to the synthetase by covalent linkage-thereby eliminating side product formation caused by diffusion. The catalytic entities responsible for the incorporation of a distinct building block into the product are called modules. Each module carries out several chemical steps required for the synthesis of nonribosomal peptides: Recognition of the building block, activation, covalent attachment, translocation, and condensation. In several cases, additional modifications are found, such as epimerization [Tyrocidine, (5)], cyclization [Gramicidin S, (6)], oxidation [Epothilone, (7)], reduction [linear gramicidin, (8)], methylation [Cyclosporin, (9)], and formylation [linear gramicidin, (10)]. In vitro studies have shown that each module can be subdivided into catalytically active domains to which the different reactions mentioned above can be assigned (1-4). Thus, the so-called adenylation (A), peptidyl carrier protein (PCP), and the condensation (C) domains were identified as being essential to all NRPSs. In addition, a second group of so-called optional domains exists: the epimerization (E), cyclization (TE or Cy), oxidation (Ox), reduction (R), N-methylation (Mt), and formylation (F) domains. Aside from NRPSs themselves, several enzymes are known to act on some peptides while they are still bound to the synthetase or even after their release. These modifying enzymes can glycosylate [Vancomycin, (11)], halogenate [Vancomycin, (11)], or reduce [linear gramicidin, (8)] the peptides in trans. With several hundred different building blocks found in nonribosomal peptide products, it becomes evident that their diversity is vast (Fig. 2). This article addresses the biologic background of these secondary metabolites, the enzymatic machineries of NRPSs, and the chemical reactions catalyzed by their domains. Furthermore, the possibility of manipulating NRPSs and using certain domains to produce novel compounds is discussed.

Figure 1. Comparison of ribosomal and nonribosomal peptide synthesis. (a) In the ribosomal information pathway, the sequence of codons in the mRNA determines the sequence of amino acids in the protein, whereas (b) the sequence of modules in the nonribosomal peptide synthetases intrinsically determines the primary sequence of the peptide product.

Figure 2. A selection of nonribosomal peptides. Chemical and structural features that contribute to the vast diversity of this class of metabolites are highlighted: Heterocycle (bacitracin), lactone (surfactin, daptomycin), ornithine and lactam (Tyrocidine), sugar, chlorinated aromats, C-C crosslink (Vancomycin), N-formyl groups (Coelichelin and linear gramicidin), fatty acid (daptomycin), dihydroxybenzoate and trimeric organization (bacillibactin), dimeric organization (gramicidin S), and ethanolamine (linear gramicidin).
Biologic Background
Nonribosomal peptides are produced by a large number of bacteria, fungi, and lower eucaryotes. For most of these compounds, their biologic role is unknown. One might suspect that these secreted molecules are used for unknown forms of communication or simply to critically increase the chance of survival for the producing cell in its habitat, because the metabolic cost of their production is enormous. However, the function of some nonribosomal compounds has been identified: The well-studied penicillin produced by Penicillium notatum, for instance, is a weapon against nutrient competitors, and the siderophore bacil- libactin helps its producer, Bacillus subtilis, acquire iron and thereby prevent iron starvation. For us, natural products produced by microorganisms attract considerable attention because their observed bioactivities range from antibiotic to immunosuppressive and from cytostatic to antitumor. Not only have these secondary metabolites been optimized for their dedicated function over millions of years of evolution, but they also represent promising scaffolds for the development of novel drugs with improved or altered activities.
Catalytic Domains of Nonribosomal Peptide Synthetases
The catalytically active entities that NRPSs are composed of can be classified as being essential to all NRPSs or being responsible for special modifications. Only when a set of domains correctly acts in appropriate order, the designated product can be synthesized (3) (Fig. 3). The function, chemistry, and interactions of these domains are discussed in the following section (Fig. 4).
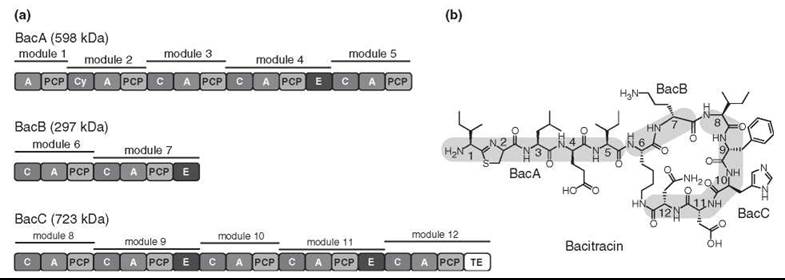
Figure 3. The nonribosomal machinery (a) needed to produce bacitracin (b) consists of three separate synthetases: BacA, BacB, and BacC, composing a total of 12 modules and 40 catalytic domains. These synthetases specifically interact with each other to produce the nonribosomal peptide bacitracin.

Figure 4. Single reactions in NRPSs and their timing. (a) After ribosomal synthesis of the opo-enzymes, the PCP domains are postsynthetically modified with 4'-Phosphopantetheine cofactors by a 4'Ppan transferase, e.g., Sfp. (b) In a second step, the A domains bind their cognate substrates as well as ATP and form the corresponding acyl-adenylate intermediates. These are transferred onto the cofactor of the neighboring PCP domains. (c) The C domains catalyse the condensation of two building blocks. The specificities of C domains and the affinities of aminoacyl-/peptidyl-ho/o-PCP domains ensure that no internal start reactions occur: (d) Only after the first condensation domain has acted does the second C domain seem to process the intermediate. During synthesis, the growing product chain is continuously translocated toward the C-terminal end of the enzyme.
Essential domains of NRPSs
Before any peptide formation can occur, the amino acids or, generally speaking, the building blocks to be condensed need to be recognized and activated (12). The adenylation (A) domains are capable of specifically binding one such building block. Once bound, the same enzyme catalyses the formation of the corresponding acyl-adenylate-monophosphate by consumption of ATP (Fig. 4). The resulting mixed anhydride is the reactive species that can be processed additionally by the NRPS machinery. Sequence alignments, mutational studies, and structural data have revealed that amino acid residues at certain positions in the enzyme determine the specificity of an A domain (13).
This result can be explained by the thereby generated chemical and physical environment of the substrate binding site. Some A domains, however, are known to have a relaxed substrate specificity. In these cases, chemically or sterically similar amino acids also are recognized, processed analogously, and thus found at that very position in the product. For example, the A domain of the first module of the gramicidin synthetase LgrA (10) not only activates valine but also activates isoleucine, which is found in 5% of linear gramicidins extracted from producing strains.
When the first building block has been recognized and activated by the A domain, the next essential domain comes into play: The peptidyl carrier protein (PCP) domain. Like the acyl carrier protein (ACP) in fatty acid biosynthesis, this PCP domain is responsible for keeping the reaction intermediates bound to the enzymatic machinery. Thus, a directed order of additional reaction steps can be implemented by controlled translocation, and NRPSs are thus often described as assembly line-like machineries. The PCP domain consists of 90 amino acid residues, roughly, and is known to rearrange itself to at least three different tertiary structures in aqueous solution, as is necessary for interaction with the surrounding domains at certain stages of synthesis (14). Just like ACPs, the PCP domains are also dependent on a post-translational modification to function. This modification is the attachment of a 4'-phosphopantetheine cofactor to a conserved serine residue. The terminal thiol group of this cofactor is the nucleophile that attacks the mixed anhydride (acyl-AMP) and therefore covalently binds the NRPS substrates via a thioester bond. After such an acylation, the PCP domain directs the substrate toward the next processing domain. If we leave out any optional modifying domains at this point, this next domain would generally be a condensation (C) domain.
The C domain is needed for the condensation of two biosynthetic intermediates during nonribosomal peptide assembly (15). The PCP-bound electrophilic donor substrate is presented from the N-terminal side of the synthetase. On the other side, the nucleophilic acceptor substrate-bound analogously to the PCP domain of the next module-reaches back to the active site of the C domain from the other direction (downstream). In the first condensation reaction of an NRPS, both of these substrates would typically be aminoacyl groups connected to their PCP domains. Condensation is initiated by the nucleophilic attack of the a-amino group of the acceptor substrate onto the thioester group of the donor substrate. The cofactor of the upstream PCP domain is released, and the resulting amide bond now belongs to the dipeptide, which remains bound to the downstream PCP domain. Thus, a translocation of the condensation product toward the next module has occurred. All condensation reactions are strictly unidirectional-always transporting the growing product chain toward the module closer to the C-terminus of the machinery. The elongated peptide then serves as the donor in a subsequent condensation step on the next module. Usually, there are as many condensation domains in an NRPS as there are peptide bonds in the linear peptide product. This general translocation model implies that the biosynthesis is linear— altogether dependent on delicate, situationally changing affinities that guarantee correct timing for each reaction and that prevents side reactions (Fig. 4). Even though this model successfully puts the biosynthetic enzymes in relation with their products for most known NRPS systems, some exceptions are known: The structures of syringomycin (16) or coelichelin (17) cannot be sufficiently explained by merely deciphering the buildup of their NRPSs when using this model. Obviously, other regulatory mechanisms and forms of inter-domain communication are not yet fully understood.
When the last condensation reaction has occurred, the linear precursor needs to be released from the enzyme. For this important last step, several mechanisms are known: simple hydrolysis of the thioester (balhimycin, vancomycin), intramolecular cyclization leading to a lactam (tyrocidine, bacitracin) or a lactone (surfactin), or even reductive thioester cleavage (linear gramicidin). In some cases, the linear precursor is dimerized (gramicidin S) or even trimerized (bacillibactin, enterobactin) before cyclization (Fig. 2). Even though these reactions are critical for the compound’s bioactivity, the catalytic domains responsible for the release are not found in all NRPS systems and will therefore be called “modifying” domains.
Modifying domains of NRPSs
Apart from the essential domains in NRPSs, several so-called modifying domains are not found in every NRPS system. Nevertheless, they are required for proper processing of their designated substrate within their synthetase. Deletion or inactivation of these modifying domains usually results in the production of compounds with bioactivities severely reduced or altogether abolished.
Most nonribosomal peptides have a cyclic connectivity. In these cases, a C-terminal, so-called thioesterase (TE) domain, is often found in the synthetase. These TE domains all share an invariant serine residue belonging to a catalytic triad (Asp-His-Ser), which is known to be acylated with the linear peptide before cyclization (18). Once the substrate is translocated from the PCP domain onto the TE domain, the regiospecific and stereospecific intramolecular attack of a nucleophile onto the C-terminal carbonyl group of the substrate is directed by the enzyme. This nucleophile can be the N-terminal α-amino group of the linear peptide (tyrocidine, gramicidin S), a side-chain amino (bacitracin) or hydroxyl group (surfactin). Since the ester bond between the substrate and the TE domain is cleaved by these cyclization reactions, the resulting lactams or lactones are released from the synthetic machinery by this step. In a few cases, the modular arrangement of NRPSs suggests that only one half (gramicidin S) or one third (bacillibactin, enterobactin) of the extracted peptide product can be produced by one assembly line-like synthesis (Fig. 2). These synthetases are considered iterative (19) because they have to complete more than one linear peptide synthesis before one molecule of the secondary metabolite can be released. According to a proposed model, the first precursor is translocated onto the TE domain, the second monomer is then produced and transferred to the TE domain-bound first monomer leading to a dimer. An analogous trimerization occurs—if applicable—and finally the product is released by cyclization.
Another modifying reaction that is commonly found in NRPSs is the epimerization (E) of an amino acid (5). E domains that are always situated directly downstream of a PCP domain catalyse these reactions. The most C-terminal amino acid of the reaction intermediate is racemized by an E domain, no matter whether the substrate is an aminoacyl group alone or a peptidyl group. The mechanism of these E domains is so far unclear, even though a catalysis that involves one or more catalytic bases to deprotonate the a-carbon atom as a first step seems likely. The resulting planar double-bond species then needs to be repro- tonated from the other side to invert the absolute configuration of the building block. This result can be accomplished by a nearby protonated catalytic base in the enzyme or water, which is positioned opposite of the first catalytic base. Nevertheless, a mixture of both stereoisomers always can be detected when the substrate bound to the enzyme is analyzed, which is indicative for either a nonstereoselective or a reversible reaction. Once the epimerized substrate undergoes the subsequent condensation reaction, only the species with an inverted stereocenter is found in the elongated product. Thus, the C domain only processes the inverted species. In some rare cases, the C domain also exhibits epimerization activity besides its normal function, and it is then called the “dual C/E” domain [Arthrofactin, (20)].
Another structural feature often found in NRPS products is N-methylated amide bonds. The domain that introduces this C1 unit, the so-called methyltransferase (Mt) domain, is situated between the A and the PCP domain (21). By consumption of S-Adenosyl-methionine, the α-amino group of the acceptor substrate is methylated before condensation with the donor.
In the case of linear gramicidin, the N-terminus of the nonribosomal peptide carries a formyl group (10). Just like in the bacterial ribosomal synthesis, only a formylated first building block is processed additionally by the corresponding enzymatic machinery. Thus, one can find a distinct formylation (F) domain at the very N-terminus of the synthetase. Another formylated NRPS product is coelichelin whose N-terminal ornithine residue is believed to be Nδ-formylated in trans by a formyltransferase genetically associated with the NRPS (17). Formyl-tetrahydrofolate is used as source of the formyl group by these enzymes.
The essential condensation domain mentioned above can, in some cases, not only condensate but also catalyze a side-chain cyclization. It is then called cyclization (Cy) domain. The cyclization is initiated by a nucleophilic attack of the side-chain heteroatom on the carbonyl group of the amide bond formed by the same domain. When water is eliminated, a stable pentacycle is integrated into the peptide chain without altering the rest of the backbone. The nucleophiles known to be reactants in these Cy domain reactions are either threonine/serine [mycobactin A, (22)] or cysteine [bacitracin, (23)]. The former leads to (methyl-)oxazoline heterocycles, whereas the latter gives rise to thiazoline-like units. Another domain sometimes associated with this heterocyclization is the oxidation (Ox) domain [Epothilone, (7)]. It is located between A and PCP domains, and it catalyses the oxidation of oxa/thiazoline intermediates, which leads to oxazoles or thiazoles, respectively.
Techniques for the Production of Novel Nonribosomal Peptides
With a constantly growing number of pathogenic bacterial strains resistant to the known antibiotics, the demand for novel antibiotics or, more generally speaking, therapeutic agents is evident. Because many NRPS products already have such activities and their chemical and structural diversity is so huge, efforts have been made to use NRPSs to broaden the known spectrum of therapeutics. In this section, the possibilities of using NRPS machineries or parts of them to produce new bioactive compounds are addressed.
Module or domain exchange
When considering the modular buildup of NRPSs, the possibility of altering the peptide product by insertion, deletion, or exchange of modules seems to be an obvious approach for the production of new compounds. Because the A domains determine the specificity of each module, even an exchange of fractions smaller than whole modules in a synthetase could lead to an altered product. In the past, various attempts have succeeded using these strategies (24, 25). For instance, the exchange of an A domain in the surfactin NRPS with other A domains of both bacterial and fungal origin lead to the formation of the expected variants of surfactin (26). However, in all of these early studies, the apparent turnover rates were significantly lower than in the wild-type systems. According to common understanding of NRPSs, two explanations for the drastically slowed down synthetic process can be given. First, the borders chosen to dissect and to fuse the catalytic domains might have been unsuitable. Even though the reoccurring, highly variable so-called linker regions between each pair of catalytic domains seem not to exhibit secondary structures, their sequence and length might be critical for proper inter-domain communication. So far, no structure of any enzyme consisting of two or more NRPS domains has been published, which makes it difficult to define the right domain border when preparing a cloning strategy for fusion or for dissection. Second, the specificity of the C domains might result in a reduced product turnover. Even though a relaxed specificity for the donor substrate has been reported, the acceptor site seems to be highly specific, which discriminates against artificial substrates (27). Both the mode of catalytic action and the molecular and structural basis for the selectivity are not fully understood for C domains so that a straightforward approach for overcoming these low turnover rates currently cannot be given.
Changing the specificity code for A domains
Sequence alignments of A domains have revealed that domains activating the same type of building block share a set of conserved residues in the primary protein sequence (13). With the A domain’s crystal structure, one can find that these residues form the substrate binding pocket (28). These residues are therefore referred to as the “selectivity-conferring code” of NRPSs (13). One can now rationally exchange these sets of residues and can obtain fully functional A domains with altered substrate recognition. For example, this process has been done for the first module of the surfactin synthetase srfAA, which activated glutamate (29). In this case, the only difference in the selectivity-conferring code compared with a glutamine activating A domain lies in one residue. Thus, the single mutation of Lys239 to Gln239 in the enzyme leads to the desired and predicted shift of specificity. In another experiment, three residues were altered to change the substrate recognition of the aspartate activating A domain in srfB2 to asparagine. The corresponding bacterial strain was shown to produce the expected variant of surfactin containing asparagine at position 5. Even though this elegant way of manipulating NRPSs works, a few drawbacks are worth mentioning. On the one hand, product turnover rates are predicted to be very low. As discussed, the C domains that have to process the artificial substrates are predicted to discriminate against non-natural substrates, which kinetically impede product formation drastically. On the other hand, this method is limited to the building blocks that other known A domains activate. Yet, the vast diversity and bioactivity of nonribosomal peptides mainly arises from their unusual connectivities and a large number of postsynthetic modifications, which one cannot address when merely changing the A domains’ specificities.
Chemoenzymatic approaches
A very powerful method for producing novel antibiotics is the chemoenzymatic approach (30). The idea behind this strategy is to leave out the enzymatic buildup of the linear peptide scaffolds and replace it by solid-phase peptide synthesis (Fig. 5). Once the desired peptide is produced, its C terminus needs to be synthetically activated (usually as a thioester) before the substrate can be subjected to enzymatic cyclization using a TE domain. The advantages of solid-phase synthesis (SPPS) are obvious: Virtually any oligo-peptide can be made in a short time and in large quantities. Even though this is true for most oligo-peptides, some amino acid sequences seem very difficult to synthesize, and the popular Fmoc protective group strategy always imposes the risk of racemization. Many different building blocks (already modified with protective groups necessary for SPPS) can be purchased, and by automated parallel peptide synthesis whole libraries can be produced very quickly. The reason why one can use such peptides as substrates lies in the relaxed substrate specificity of many TE domains. The TE domain of the Tyrocidine synthetase, which carries out a head-to-tail cyclization of the de- capeptide DPhe-Pro-Phe-DPhe-Asn-Gln-Tyr-Val-Orn-Leu, for instance, only recognizes the two N-terminal and C-terminal amino acids of the natural substrate. The side chains of the amino acids at other positions are not recognized by the enzyme, and experiments with substrates that carry substitutions to alanine in these positions still lead to analogous cyclodecamers (31). The major advantage of using TE domains for cyclization reactions is their regiospecificity, stereospecificity, and chemo- specificity. Thus, no protective groups are needed during these enzymatic cyclization reactions, and undesired side product formation is minimized. Additionally, these reactions are carried out under mild aqueous conditions, usually pH 7-8.

Figure 5. Chemoenzymatic approaches for the production of novel bioactive compounds. In this example, the enzymatic buildup of the linear precursor of daptomycin by its NRPSs (DptA, DptBC, and DptD) is substituted by solid-phase synthesis (a). By using the 4'Ppan transferase Sfp and the CoA-thioester of the linear peptide, the opo-enzyme PCP-TE and be modified, and after trans-esterification cyclized by the TE domain (b). Because the resulting holo-enzyme cannot be modified again, this is a single turnover reaction. Another strategy uses thiophenole-esters of the linear peptides to be cyclized (c). When these compounds are used, no PCP domain is necessary. The TE domain is readily acylated, and regiospecific and stereospecific cyclization toward daptomycin or, depending on the linear peptide provided, toward variants thereof occurs. Because the enzyme is not altered in any way after product release, this setup results in a multiple turnover.
To follow this chemoenzymatic approach, the synthetic substrates must be transferred onto the catalytically active serine residue of the TE domain. This transfer can either be done directly or with the help of a PCP domain. In the natural system, translocation is realized by the interaction between the PCP and the TE domain. The substrate, which is bound to the 4'Ppan cofactor of the PCP domain as a thioester, acylates the hydroxyl group of the serine. Chemically speaking, the acylation of the TE is a result of a trans-esterification. When using TE domains, the substrate provided in trans must also have an appropriate acylation potential. Several key techniques have been developed to covalently attach synthetic substrates to PCP and TE domains. In the first method, the relaxed substrate specificity of the 4'Ppan transferase Sfp is used to load acyl moieties onto PCP domains enzymatically. Just like in the natural priming reaction in which the 4'Ppan part of CoA is transferred onto the conserved serine residue of the apo-PCP domain, Sfp does analogously attach S-acylated 4'Ppans, which originates from S-acylated CoA substrates (32). These CoA substrates can readily be obtained by one coupling reaction directly after solid phase synthesis. With this technique, virtually any substrate can be brought to a desired position in recombinant NRPS enzymes containing an apo-PCP domain. This result is of great value when elucidating the catalytic properties and substrate specificities of other domains. When investigating TE domains, for example, the corresponding apo-PCP-TE would be the starting point for screening the cyclization abilities of the TE domain with a synthetic substrate library. However, the major disadvantage of this method becomes evident when looking closely at the enzyme after the release of the product: The PCP domain is now in its holo-state, and therefore, subsequent enzymatic loading of substrates with Sfp is impossible. Because product formation is limited to a single turnover, other methods have been developed to allow for multiple turnover.
For multiple turnover reactions, the TE domains must be supplied with substrates in trans; yet the acylation potential must be sufficient and the compound must be recognized by the enzyme. The first approach made was inspired by the natural system where the substrate is activated as a thioester bound to the 4'Ppan cofactor. The idea was to minimize the 4'Ppan moiety by replacing it with N-acetyl-cysteamine (SNAc) (33). This method works fairly well; however, it seems in later studies that the acylation potential is of greater importance than the similarity to the natural situation and a variety of peptidyl-thioesters with better leaving groups than SNAc was tested. The fastest turnover rates were found when thiophenol-esters were used (30, 34). Thiophenol has several advantages: It does not have any functional groups other than the thiol group, it is inexpensive, and it can easily be separated from the product. This method has been successfully used to shed light on the promiscuity of the TE domain of the daptomycin NRPS (35).
Even though these techniques allow for the production of new potentially bioactive compounds, they are usually closely related to known substances, which basically implies an analogous mode of action, and the deviations normally alter quantitative parameters such as solubility and affinity. Nevertheless, hundreds of nonribosomal systems still need to be explored, and the discoveries of new ones are frequently reported. The basic understanding of the catalytic functions of nonribosomal domains and modules that we have today is a good starting point for additional exploration and use of systems that are not yet understood fully.
References
1. Walsh CT. Polyketide and nonribosomal peptide antibiotics: modularity and versatility. Science 2004; 303:1805-1810.
2. Finking R, Marahiel MA. Biosynthesis of nonribosomal peptides1. Annu. Rev. Microbiol. 2004; 58:453-488.
3. Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem. Rev. 2006; 106:3468-3496.
4. Keller U, Schauwecker F. Combinatorial biosynthesis of nonribosomal peptides. Comb. Chem. High Throughput Screen. 2003; 6:527-540.
5. Stein DB, Linne U, Hahn M, Marahiel MA. Impact of epimerization domains on the intermodular transfer of enzyme-bound intermediates in nonribosomal peptide synthesis. ChemBioChem. 2006; 7:1807-1814.
6. Kohli RM, Trauger JW, Schwarzer D, Marahiel MA, Walsh CT. Generality of peptide cyclization catalyzed by isolated thioesterase domains of nonribosomal peptide synthetases. Biochemistry 2001; 40:7099-7108.
7. Chen H, O’Connor S, Cane DE, Walsh CT. Epothilone biosynthesis: assembly of the methylthiazolylcarboxy starter unit on the EpoB subunit. Chem. Biol. 2001; 8:899-912.
8. Schracke N, Linne U, Mahlert C, Marahiel MA. Synthesis of linear gramicidin requires the cooperation of two independent reductases. Biochemistry. 2005; 41(44):8507-8513.
9. Velkov T, Lawen A. Mapping and molecular modeling of S-adenosyl-L-methionine binding sites in N-methyltransferase domains of the multifunctional polypeptide cyclosporin synthetase. J. Biol. Chem. 2003; 278:1137-1148.
10. Schoenafinger G, Schracke N, Linne U, Marahiel MA. Formylation domain: an essential modifying enzyme for the nonribosomal biosynthesis of linear gramicidin. J. Am. Chem. Soc. 2006; 128:7406-7407.
11. Hubbard BK, Walsh CT. Vancomycin assembly: nature’s way. Angew. Chem. Int. Ed. Engl. 2003; 42:730-765.
12. Luo L, Burkart MD, Stachelhaus T, Walsh CT. Substrate recognition and selection by the initiation module PheATE of gramicidin S synthetase. J. Am. Chem. Soc. 2001; 123:11208-11218.
13. Stachelhaus T, Mootz HD, Marahiel MA. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 1999; 6:493-505.
14. Koglin A, Mofid MR, Lohr F, Schafer B, Rogov VV, Blum MM, et al. Conformational switches modulate protein interactions in peptide antibiotic synthetases. Science. 2006; 14(312):273-276.
15. Keating TA, Marshall CG, Walsh CT, Keating AE. The structure of VibH represents nonribosomal peptide synthetase condensation, cyclization and epimerization domains. Nat. Struct. Biol. 2002; 9:522-526.
16. Bender CL, Alarcon-Chaidez F, Gross DC. Pseudomonas syringae phytotoxins: mode of action, regulation, and biosynthesis by peptide and polyketide synthetases. Microbiol. Mol. Biol. Rev. 1999; 63:266-292.
17. Lautru S, Deeth RJ, Bailey LM, Challis GL. Discovery of a new peptide natural product by Streptomyces coelicolor genome mining. Nat. Chem. Biol. 2005; 1:265-269.
18. Kohli RM, Trauger JW, Schwarzer D, Marahiel MA, Walsh CT. Generality of peptide cyclization catalyzed by isolated thioesterase domains of nonribosomal peptide synthetases. Biochemistry 2001; 40:7099-7108.
19. Gehring AM, Mori I, Walsh CT. Reconstitution and characterization of the Escherichia coli enterobactin synthetase from EntB, EntE, and EntF. Biochemistry 1998; 37:2648-2659.
20. Balibar CJ, Vaillancourt FH, Walsh CT. Generation of D amino acid residues in assembly of arthrofactin by dual condensation/epimerization domains. Chem. Biol. 2005; 12:1189-1200.
21. Billich A, Zocher R. N-Methyltransferase function of the multifunctional enzyme enniatin synthetases. Biochemistry 1987; 26:8417-8423.
22. Quadri LE, Sello J, Keating TA, Weinreb PH, Walsh CT. Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem. Biol. 1998; 5:631-645.
23. Eppelmann K, Doekel S, Marahiel MA. Engineered biosynthesis of the peptide antibiotic bacitracin in the surrogate host Bacillus subtilis. J. Biol. Chem. 2001; 276:34824-34831.
24. Mootz HD, Kessler N, Linne U, Eppelmann K, Schwarzer D, Marahiel MA. Decreasing the ring size of a cyclic nonribosomal peptide antibiotic by in-frame module deletion in the biosynthetic genes. J. Am. Chem. Soc. 2002; 124:10980-10981.
25. Schauwecker F, Pfennig F, Grammel N, Keller U. Construction and in vitro analysis of a new bi-modular polypeptide synthetase for synthesis of N-methylated acyl peptides. Chem. Biol. 2000; 7:287-297.
26. Stachelhaus T, Schneider A, Marahiel MA. Rational design of peptide antibiotics by targeted replacement of bacterial and fungal domains. Science 1995; 269:69-72.
27. Belshaw PJ, Walsh CT, Stachelhaus T. Aminoacyl-CoAs as probes of condensation domain selectivity in nonribosomal peptide synthesis. Science 1999; 284:486-489.
28. May JJ, Kessler N, Marahiel MA, Stubbs MT. Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:12120-12125.
29. Eppelmann K, Stachelhaus T, Marahiel MA. Exploitation of the selectivity-conferring code of nonribosomal peptide synthetases for the rational design of novel peptide antibiotics. Biochemistry. 2002; 41:9718-9726.
30. Grunewald J, Marahiel MA. Chemoenzymatic and template-directed synthesis of bioactive macrocyclic peptides. Microbiol. Mol. Biol. Rev. 2006; 70:121-146.
31. Trauger JW, Kohli RM, Mootz HD, Marahiel MA, Walsh CT. Peptide cyclization catalysed by the thioesterase domain of tyrocidine synthetase. Nature 2000; 407:215-218.
32. Sieber SA, Walsh CT, Marahiel MA. Loading peptidyl-coenzyme A onto peptidyl carrier proteins: a novel approach in characterizing macrocyclization by thioesterase domains. J. Am. Chem. Soc. 2003; 125:10862-10866.
33. Ehmann DE, Trauger JW, Stachelhaus T, Walsh CT. Aminoacyl-SNACs as small-molecule substrates for the condensation domains of nonribosomal peptide synthetases. Chem. Biol. 2000; 7:765-772.
34. Sieber SA, Tao J, Walsh CT, Marahiel MA. Peptidyl thiophenols as substrates for nonribosomal peptide cyclases. Angew. Chem. Int. Ed. Engl. 2004; 43:493-498.
35. Grunewald J, Sieber SA, Mahlert C, Linne U, Marahiel MA. Synthesis and derivatization of daptomycin: a chemoenzymatic route to acidic lipopeptide antibiotics. J. Am. Chem. Soc. 2004; 126:17025-17031.
Further Reading
Baltz RH. Molecular engineering approaches to peptide, polyketide and other antibiotics. Nat. Biotechnol. 2006; 24:1533-1540.
Cane DE. Introduction: Polyketide and Nonribosomal Polypeptide Biosynthesis. From Collie to Coli. Chem. Rev. 1997; 97:2463-2464.
Du L, Shen B. Biosynthesis of hybrid peptide-polyketide natural products. Curr. Opin. Drug Discov. Devel. 2001; 4:215-228.
Grunewald J, Kopp F, Mahlert C, Linne U, Sieber SA, Marahiel MA. Fluorescence resonance energy transfer as a probe of peptide cyclization catalyzed by nonribosomal thioesterase domains. Chem. Biol. 2005; 12:873-881.
Hahn M, Stachelhaus T. Selective interaction between nonribosomal peptide synthetases is facilitated by short communication-mediating domains. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:15585-15590.
Salomon CE, Magarvey NA, Sherman DH. Merging the potential of microbial genetics with biological and chemical diversity: an even brighter future for marine natural product drug discovery. Nat. Prod. Rep. 2004; 21:105-121.
Sieber SA, Linne U, Hillson NJ, Roche E, Walsh CT, Marahiel MA. Evidence for a monomeric structure of nonribosomal Peptide synthetases. Chem. Biol. 2002; 9:955-956.
See Also
Antibacterial Drugs, Design of
Antibiotics From Microorganisms
Antibiotics, Synthesis of
Antibiotics, Mechanism of Action
Bacterial Resistance to Antibiotics
Coupling Methods: Peptide Synthesis
Natural and Unnatural Amino Acids, Synthesis of
Natural Products as Anticancer Agents
Natural Products in Microbes, Chemical Diversity of
Peptides, Chemistry of
Peptide Combinatorial Libraries
Peptide Synthesis
Pharmaceuticals, Natural Products and Natural Product Models of
Polyketides as Drugs
Polyketide Biosynthesis