CHEMICAL BIOLOGY
Post-Translational Modifications to Regulate Protein Function
Hening Lin, Jintang Du, and Hong Jiang, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York
doi: 10.1002/9780470048672.wecb467
Protein post-translational modifications (PTM) are very important to regulate protein function and to control numerous important biological processes. Here a brief review of commonly found enzyme-catalyzed PTM is given. These PTM include modifications that occur on protein side chains and those that involve protein backbones. The introduction of different PTM is followed by a summary of the molecular basis for the regulation of protein function by PTM. The focus is then given to a few major PTM that play important roles in eukaryotes, such as phosphorylation, methylation, acetylation, glycosylation, ubiquitylation, and proteolysis. For each modification, a description will be given about the residues modified, the enzymatic reaction mechanisms, the major known biological functions, and its relevance to human diseases. At the end, we discuss challenges in identifying new pathways regulated by known PTM and discovering new PTM.
The central dogma of molecular biology, DNA is transcribed to mRNA which is then translated to proteins, implies the importance of proteins. After all, it is the proteins that carry out most of the biological functions of a cell. Thus controlling transcription and translation are very important, as they ultimately control what proteins are synthesized in cells and thus control the properties of cells. However, one should not overlook what happens to proteins after they are synthesized. Many chemical modifications can occur to proteins after translation. Collectively, these modifications are called post-translational modifications (PTM). PTM are very important in regulating protein function, which is reflected by the large number of genes devoted to catalyzing PTM. For example, in the human genome (with less than 30,000 genes total), more than 500 kinases catalyze protein phosphorylation (1), and more than 500 proteases catalyze the hydrolytic cleavage of proteins (2). Deregulation in PTM is the cause of various human diseases, as will be explained later in specific PTM sections. Here, a brief review is given on different types of PTM and on how PTM regulate protein function. Some basic principles will be highlighted so that readers who are unfamiliar with PTM can have a quick but comprehensive understanding of PTM. The recent book on PTM by Professor Walsh from Harvard Medical School provides a more complete description of PTM (3). Where appropriate, references on specific PTM will also be given in different sections for additional information. The abbreviations used are cataloged in Table 1 to help readers who are not familiar with the biological language.
Types of post-translational modifications
PTM can be enzyme-catalyzed and thus controlled carefully, or they can be nonenzymatic with less control. For example, protein glycation during hyperglycemia is a nonenzymatic PTM that accounts for some symptoms of diabetes (4). Protein nitrosylation on Cys residues is another nonenzymatic PTM that can affect protein function (5). Coordination by metal ions can also be considered as a PTM. For many proteins, metal binding is crucial for maintaining the correct structure or the enzymatic activity (6). Here, the focus will be given to enzyme-catalyzed PTM. Figures 1 and 2 show many commonly found enzyme-catalyzed PTM. (3)
As can be observed from Fig. 1, most PTM happen to protein side chains. Typically, the side chains involved are nucleophilic, such as Cys (palmitoylation, isoprennylation, disulfide bond formation, ADP-ribosylation), Lys (acetylation, methylation, ubiquitinylation), Arg (methylation, ADP-ribosylation), Asp/Glu [methylation, poly(ADP-ribosyl)ation], Ser/Thr (phosphorylation, O-glycosylation), and Tyr (phosphorylation). Weaker nucleophiles are also used, such as the side chain amide nitrogen in Asn (in N-glycosylation), the C-2 position of Trp (in C-glycosylation), and the C-2 position of His (in diphthamide). In amidation reactions catalyzed by transglutaminases and polyglutamylation/polyglycylation reactions that happen to Glu residues, the ε-NH2 from Lys or α-NH2 from Glu/Gly acts as the neucleophile, whereas the side chain of Gln or Glu serves as the electrophile. In addition, several amino acid side chains can be oxidized, such as Pro, Lys, Asn, Tyr, Trp, and Cys, to give oxidized amino acids.
A few PTM reactions also involve changes in protein backbone. These reactions include the hydrolytic cleavage of the peptide backbone by proteases, the anchoring of proteins to glycosylphosphotidylinositol (GPI) or cholesterol, and the C-terminal amide formation by oxidative cleavage of glycine residues. Some PTM involve changes in both the side chain and the main chain, such as the formation of 4-methylidene-5-imiazole-5-one (MIO) prosthetic group in deaminases and aminomutases, the formation of the fluorophore in GFP (green fluorescent protein), and the formation of pyruvamide in decarboxylases (Fig. 2).

Figure 1. Major enzyme-catalyzed PTM that modify protein side chains.

Figure 2. A few PTM that involve protein backbone.
Table 1. List of abbreviations
|
ABL |
A tyrosine kinase encoded from abl (Abelson) gene, the fusion protein ABL-BCR is involved in inhibition of apoptosis in chronic myelogenous leukemia cells |
|
AC
|
Adenylate cyclase, converts ATP to cyclic AMP |
|
AcLys |
Acetyllysine |
|
ACP |
Acyl carrier protein, found in fatty acid synthases and polyketide synthases, functions to carry the elongating fatty acyl chain |
|
ADAM |
A disintergrase and metalloprotease, a family of proteases that hydrolyze off extracellular portions of transmembrane proteins |
|
ADP |
Adenosine 5’-diphosphate |
|
Apaf-1 |
Apoptotic protease activation factor-1, a cytosolic protein involved in cell death or apoptosis, interacts with cytochrome c to activate caspase 9 |
|
AT |
Acyltransferase, found in fatty acid syntheases and polyketide syntheases, adds a malonyl group to the holo form of the ACP domain |
|
ATP |
Adenosine 5’-triphosphate |
|
Bcl2 |
Named from B-cell lymphoma 2, an antiapoptotic protein |
|
BCR |
A protein encoded from breakpoint cluster region gene, has serine/threonine kinase activity. Fusion with abl protein causes leukemia |
|
cAMP |
3’-5’-cyclic adenosine monophosphate |
|
CARD |
Caspase recruitment domain, mediates the formation of larger protein complexes via direct interactions between individual cards, involved in the regulation of caspase activation and apoptosis |
|
CARM1 |
Coactivator-associated arginine(R) methyltransferase 1, methylates Arg17 and Arg26 residues on Histone H3 |
|
Cbl-b |
Ubiquitously expressed homolog of Cbl, a mammalian protein involved in cell signaling and protein ubiquitination, named after Casitas B-lineage Lymphoma |
|
CBP |
CREB binding protein, a transcriptional co-activating protein |
|
CD2 |
Cellular differentiation marker 2, a cell adhesion protein found on the surface of T cells and natural killer cells |
|
CDK |
Cell-division kinases, serine/threonine kinases, activated by association with cyclins and involved in regulation of the cell cycle, transcription and mrna processing |
|
CHD1 |
Chromodomain helicase DNA-binding protein 1, interacts with methylated Lys4 on Histone H3 |
|
CLOCK |
A protein named from circadian locomotor output cycles kaput gene, regulating circadian rhythm |
|
CML |
Chronic myelogenous leukemia, a form of leukemia characterized by the increased and unregulated growth of predominantly myeloid cells in the bone marrow and the accumulation of these cells in the blood |
|
CREB |
Camp response element binding proteins, as transcription factors, bind to certain sequences called camp response elements (CRE) in DNA and thereby increase or decrease the transcription of certain genes |
|
Cyto c |
Cytochrome c, a small heme protein associated with the inner membrane of the mitochondria and released in response to pro-apoptotic stimuli |
|
DED |
Death effector domain, a protein interaction domain found in inactive procaspases and proteins that regulate caspase activation in the apoptosis cascade |
|
DH |
Dehydratase, found in fatty acid syntheases and polyketide syntheases, dehydrates the P-OH of acyl thioester |
|
DNAse |
Deoxyribonuclease, catalyzes the hydrolytic cleavage of phosphodiester linkages in the DNA backbone |
|
ER |
Endoplasmic reticulum |
|
ER |
Enoylreductase, found in fatty acid syntheases and polyketide syntheases, reduces the enoyl of enoyl thioester to the saturated thioester |
|
ERK |
Extracellular signal-regulated kinase, activates many transcription factors and some downstream protein kinases, involved in functions including the regulation of meiosis, mitosis, and postmitotic functions in differentiated cells |
|
Factor IX |
One of the serine proteases of the coagulation system |
|
FAD |
Flavin adenine dinucleotide |
|
FADD |
Fas-associated protein with death domain, connects the Fas-receptor and other death receptors to caspase-8 through its death domain to form the death inducing signaling complex during apoptosis |
|
FHA domain |
Forkhead-associated domain, a phosphospecific protein-protein interaction motif involved in checkpoint control of the cell cycle |
|
Gcn5 |
A yeast transcriptional adaptor that has histone acetyltransferase activity |
|
GDP |
Guanosine-5’-diphosphate |
|
GFP |
Green fluorescent protein |
|
GPCR |
G protein-coupled receptor, a transmembrane receptor that senses molecules outside the cell and activates inside signal transduction pathways and cellular responses |
|
GPI |
Glycosylphosphatidylinositol, a glycolipid that can be attached to the C-terminus of a protein during post-translational modification |
|
GPK |
Glycogen phosphorylase kinase, a serine/threonine-specific protein kinase which activates glycogen phosphorylase by phosphorylation |
|
Grb2 |
Growth factor receptor-bound protein 2, an adaptor protein involved in signal transduction/cell communication |
|
[-2pt] GTP |
Guanosine-5’-triphosphate |
|
HDACs |
Histone deacetylases, remove acetyl groups from an e-N-acetyl lysine residues on histones |
|
hDOT1L |
Human DOTl-like protein, methylates histone H3 at Lys79. (DOT1: Yeast disruptor of telomeric silencing-1) |
|
HECT |
Homologous to E6-AP C terminus, mediates E2 binding and ubiquitination |
|
HIF |
Hypoxia inducible factor, a transcription factor that responds to changes in available oxygen in the cellular environment, specifically to decreases in oxygen or hypoxia |
|
hnRNPs |
Hetergenous nuclear ribonucleoproteins, which forms complex with pre-MRNA and MRNA and shuttles between the nucleus and the cytoplasm |
|
HP1 |
Heterochromatin protein 1, binds to heterochromatin and interacts with numerous partner proteins to organize the higher-order structure of heterochromatin |
|
IgG |
Immunoglobulin G, one antibody isotype |
|
IKK |
Inhibitor of NF-Kb kinase, which phosphorylates inhibitor of NF-Kb for the proteasomal degradation to release NF-Kb dimers to translocate to the nucleus and activate transcription of target genes |
|
IP7 |
Inositol pyrophosphate, a proposed physiologic phosphate donor |
|
JHDM |
Jmjc domain-containing histone demethylase |
|
JmjC |
Jumonji domain-containing, a novel demethylase signature motif |
|
KR |
Ketoreductase, found in fatty acid syntheases and polyketide syntheases, reduces the β-ketoacyl thioester |
|
KS |
Ketosynthase, found in fatty acid syntheases and polyketide syntheases, carries out C-C bond-forming chain elongation step |
|
LSD1 |
Lysine-specific demethylase 1, demethylates histone H3 at lysine 9 |
|
MAP Kinase or MAPK |
Mitogen-activated protein kinase, serine/threonine-specific protein kinases that respond to extracellular stimuli (mitogens) and regulate various cellular activities, such as gene expression, mitosis, differentiation, and cell survival/apoptosis |
|
Me2Lys |
ε-N-dimethyllysine |
|
Me3Lys |
ε-N-trimethyllysine |
|
MEK |
MAPK/ERK kinase, activates a MAP kinase or ERK through phosphorylation |
|
MeLys |
ε-N-monomethyllysine |
|
MIO |
4-methylidene-5-imiazole-5-one |
|
MOZ |
Monocytic leukemia zinc finger protein, a histone acetyltransferase implicated in leukemogenic and other tumorigenic processes, regulates expression of genes required for proliferation and repopulation of potential of stem cells in the hematopoietic compartment |
|
NAD |
Nicotinamide adenine dinucleotide |
|
NGFp1 |
Nerve growth factor P1, a secreted protein which induces the differentiation and survival of particular target neurons, belonging to neurotrophins protein family |
|
OSTase |
Oligosacchryltransferase |
|
PADs |
Protein Arg deiminases, hydrolyzes the guanidine side chain of Arg residues to citrulline residues in proteins |
|
PARP-1 |
Poly(ADP-ribose) polymerase-1, catalyzes the transfer of poly ADP-ribose to substrate proteins by using NAD as substrate, involved in cellular response to DNA damage and DNA metabolism |
|
PKA |
Protein kinase A, a family of kinases whose activity are dependent on the level of cyclic AMP, involved in the regulation of glycogen, sugar, and lipid metabolism |
|
PRMT |
Protein Arg(R) methyltransferase, catalyzes the transfer of methyl group from S-adenosylmethionine to the guanidino nitrogen atoms of arginine residues |
|
pSer |
Phosphoserine |
|
PTB |
Phosphotyrosine binding |
|
pThr |
Phosphothreonine |
|
PTM |
Post-translational modifications |
|
PTyr |
Phosphotyrosine |
|
RAIDD |
RIP-associated ICH-1/CED-3 homologous protein with a death domain, functions as an adaptor in recruiting the death protease ICH-1 to the TNFR-1 signaling complex (ICH: Ice and ced-3 homolog; TNRF: tumor necrosis factor receptor) |
|
RING |
Really interesting new gene. Ring proteins are components of ubiquitin e3 enzyme complexes. |
|
RIP |
Receptor-interacting protein |
|
SAH |
S-adenosylhomocysteine |
|
SAHA |
Vorinostat, suberoylanilide hydroxamic acid, brand name Zolinza, a class of agents known as histone deacetylase inhibitors, as a drug for the treatment of cutaneous T cell lymphoma (a type of skin cancer) |
|
SAM |
S -adenosylmethionine |
|
SCF |
Skp1-Cullin-F Box, a multi-protein complex catalyzing the ubiquitylation of proteins destined for proteasomal degradation |
|
SET |
Supressor of variegation-Enhanser of zeste-Trithorax. SET domains have methyltranferase activity. |
|
Set8 |
A novel human SET domain-containing protein, which specifically methylates H4 at Lys20 |
|
Set9 |
A novel human SET domain-containing protein, which specifically methylates H3 at Lys4 |
|
SH2 |
Src homology 2, a phosphotyrosine-recognition protein domain of about 100 amino acid residues first identified as a conserved sequence region among the oncoproteins Src and Fps |
|
SMAD |
Proteins homologs of both the drosophila protein, mothers against decapentaplegic (MAD) and the C. Elegans protein SMA, as signal-activated transcription factors regulated by the TGF-β superfamily |
|
Smyd |
Proteins containing SET and MYND domain. MYND encoded mynd (myosin) gene, which have histone methyltransferase activity |
|
snRNPs |
Small nuclear ribonucleoproteins, combining with pre-MRNA and various proteins to form spliceosomes to removes introns from pre-MRNA segment |
|
Sos |
Son of sevenless, a guanine nucleotide exchange factor that activates Ras |
|
STAT |
Signal transducers and activators of transcription, proteins which are involved in the development and function of the immune system |
|
SUMO |
Small ubiquitin-like modifier, a family of small proteins that are covalently attached to and detached from other proteins in cells to modify their functions |
|
TAF10 |
TATA box-binding protein-associated factor 10, a component of the general transcription factor complex TFIID and the TATA box-binding protein (TBP)-free TAF-containing complex |
|
TIF2 |
Transcription intermediary factor 2, a transcriptional coregulatory protein which contains several nuclear receptor interacting domains and an intrinsic histone acetyltransferase activity |
|
TF |
Transcription factor, a protein that binds to specific region of DNA by DNA binding domains and mediates the transcription from DNA to RNA |
|
TGFpl |
Transforming growth factor |31, a secreted protein that performs many cellular functions, including the control of cell growth, cell proliferation, cell differentiation and apoptosis, belonging to the transforming growth factor beta superfamily of cytokines |
|
TGN |
Trans Golgi network, a part of the golgi apparatus in cells |
|
TOPA |
2,4,5-trihydroxyphenylalanine |
|
TRADD |
Tumor necrosis factor receptor-associated protein with death domain, an adapter protein that recruits other proteins to the cytoplasmic TNF (tumor necrosis factor) receptor complex, involved in apoptosis |
|
UAP |
Ubiquitin activating protein |
|
UBA domain |
Ubiquitin binding associated domain, one class of ubiquitin binding domains |
|
UBD |
Ubiquitin binding domain, which binds mono- or poly-ubiqitin |
|
UBP |
Ubiquitin-specific protease, hydrolyzes both linear and branched Ub modifications |
|
UDP |
Uridine diphosphate |
|
UMP |
Uridine monophosphate |
Molecular basis for the regulation of protein function by PTM
As with all other chemical species, protein structure determines protein function. PTM can regulate protein function because they can change protein structure. The structure change introduced by PTM can be local and small. For example, methylation of Lys residues makes the side chain more hydrophobic without changing protein backbone conformation significantly [at least based on crystal structures in which methylated and unmethylated histone peptides are bound by another protein (7)], whereas phosphorylation can change the backbone conformation within a limited region of a protein by charge-pairing with nearby Arg residues or by interacting with main chain NH and helical dipole (8). In contrast, some PTM can alter protein overall structure more dramatically, such as the proteolytic cleavage of proteins into smaller fragments, or the addition of protein tags like ubiquitin. These structure changes, small or big, are the basis for the biological functions of different PTM and typically lead to one or more of the consequences described below.
Changing protein structure to turn on/off catalytic activity of enzymes
The best-known PTM that is widely used to regulate enzymatic activity is phosphorylation. Phosphoryation regulates the activity of many enzymes by different mechanisms. For example, glycogen phosphorylase is activated allosterically by phosphorylation at Ser14, whereas Escherichia coli isocitrate dehydrogenase is inhibited by phosphorylation because of the block of substrate access to the active site (9). The most interesting and very important catalytic activity regulated by phosphorylation is protein kinase activity. Most protein kinases are activated by phosphorylation of Thr/Tyr residue(s) in the activation segment. The structural changes induced by phosphorylation, which are illustrated in Fig. 3 with ERK (extracellular signal-regulated kinase), convert the inactive kinases to active kinases (8). The regulation of protein kinase activity by phosphorylation bears enormous biological significance because protein phosphorylation is important in signal transduction, and the control of downstream kinase activity via phosphorylation by upstream kinase is one major method to propagate signals to downstream partners, as will be elaborated later.
Proteolysis is another way to control enzymatic activity, although unlike phosphorylation, the change in activity is irreversible. Many proteases are synthesized as inactive precursors (zymogens) that have to be cleaved by proteolysis to become active. These precursors include proteases that are secreted into digestive tracts or lysosomes, the catalytic active P subunits in the eukaryotic 20 S proteosome that are activated by self-cleavage (10), and the effector caspases involved in apoptosis that are activated by initiator caspases-mediate cleavage (11).
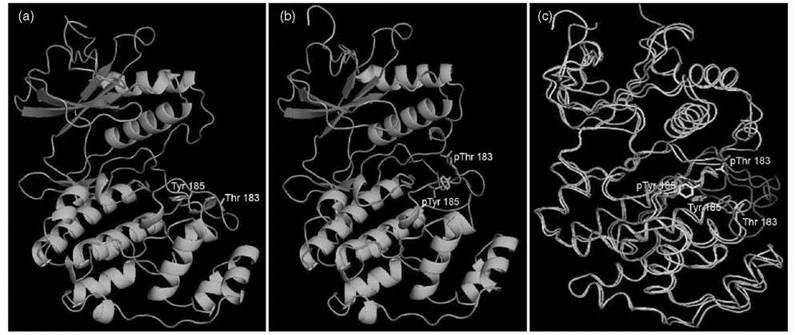
Figure 3. Structure of ERK2 in both unphosphorylated (inactive) and phosphorylated (active) state. (a) ERK2 in unphosphorylated state (figure made using PDB 1ERK); residues Thr183 and Tyr185 in the activation segment are labeled; (b) ERK2 in phosphorylated state (ERK-P2, figure made using PDB 2ERK). The two phosphorylated residues, pThr183 and pTyr185, are labeled; (c) Superposition of ERK2 and ERK2-P2.
Changing protein structure to create or to mask recognition motifs
Many PTM exert their biological functions by creating recognition motifs to recruit binding partners (12) or by masking recognition motifs to disrupt existing interactions. Phosphory- lated Ser/Thr residues can be recognized by proteins that contain 14-3-3 domains, FHA (forkhead-associated) domains, SMAD [proteins homologs of both the drosophila protein, mothers against decapentaplegic (MAD) and the Caenorhabditis elegans protein SMA] domains, and several other domains (13). Phosphorylated Tyr residues can recruit proteins that contain SH2 (Src homology 2) domains and PTB (phosphotyrosine binding) domains (14). Acetyl Lys residues can be recognized by proteins with bromodomains (15, 16), and methylated Lys residue can be recognized by proteins with chromodomains and Tudor domains (17). The ubiquitin and ubiquitin-like protein tags can also be recognized by various protein domains that mediate the biological function of modification with these protein tags (18, 19). The structures of a few domains dedicated to recognition of post-translationally modified residues are shown in Fig. 4. Typically, domains that recognize post-translationally modified residues have specificities in that they recognize not only the modified residue, but also the local structure in which the residue resides. The specific recognition of PTM in different contexts is the key to understand many biological consequences of PTM, as will be explained in more detail in particular PTM sections later.
In addition to creating recognition motifs to recruit proteins, a few PTM can also increase interaction with other species, such as the lipid bilayer of different cellular membranes. These modifications include the formation of GPI-anchored proteins (20), protein myristoylation on the a-amino group of the N-terminal Gly (21), protein C-terminal prennylation on Cys residues (22), and protein palmitoylation on Cys residues that are close to membrane surface (23). These lipid modifications occur to many signaling proteins, which include G protein-coupled receptors and small G proteins, and they play important roles in signal transduction and membrane trafficking (24).

Figure 4. Structures of a few dedicated domains that recognize post-translationally modified residues. (a) SH2 domain of v-Src in complex with pTyr peptide (pTyr-Val-Pro-Met-Leu). Residues Arg12, Arg32, Ser34, Thr36, and Lys60 from the SH2 domain interact with pTyr (figure made using PDB 1SHA); (b) Bromodomain of yeast histone acetyltransferase Gcn5 in complex with AcLys peptide (histone H4 residues 15-29, AK(Ac)RHRKILRNSIQGI). Bromodomain residues Pro351, Gln354, Tyr364, Met372, Val399, and Asn407 interact with AcLys (with some of the interaction is mediated by water molecules, figure made using PDB 1 E6I); (c) Chromodomain of HP1 in the complex with histone H3 Me3Lys9 (figure made using PDB 1KNE). Chromodomain residues Tyr 24, Trp 45, Tyr 48 and Glu 52 bind Me3Lys;(d) UBA domain of Cbl-b in complex with ubiquitin (figure made using PDB 2OOB). UBA domain residues Asp933, Ala937, Met940, Phe946, and Lys950 interact with ubiquitin residues Leu8, Ile44, Ala46, Gly47, Gln49, His68, and Val70. UBA: ubiquitin binding associated.
Adding functional groups to allow catalysis
Typically, proteins are formed with the most common 20 amino acids, which only offer a limited number of choices of functional groups for catalyzing different reactions. The limit in the number of functional groups is complemented by the use of various coenzymes or cofactors, many of which are attached covalently to the corresponding enzymes. One class of PTM with this function is the addition of “swinging arm” prosthetic groups (biotin, phosphopantetheine, and lipoic acid) to proteins (25). Biotin is used as a carrier of CO2 in carboxylation reactions, and the disulfide bond in lipoyl group is used as an electron carrier and acyl carrier in 2-keto acid dehydrogenases. The phosphopan- tetheine group provides a thiolate as the carrier of acyl chains and is used in fatty acids synthases, polyketide synthases, and nonribosomal peptide synthases (26). Although a thiolate side chain can also be provided by Cys, the longer phosphopantetheine can shuttle the acyl chains to different catalytic domains, which allows multiple reactions to occur in sequence on the acyl chains (Fig. 5). This “swinging arm” catalysis, which is also enabled by biotinylation and lipoylation, cannot be achieved by natural proteinogenic amino acids with shorter side chains.
Another type of PTM provides new functional groups for enzyme catalysis by oxidation of side chain. These include TOPA (2,4,5-trihydroxyphenylalanine) quinone in amine oxidases (Fig. 6), tryptophan tryptophanyl quinone in methylamine dehydrogenase (27) and formylglycine in sulfatases. (28) Main chain modifications can also generate prosthetic groups for enzyme catalysis, such as the MIO group in His/Phe ammonia-lyase (29, 30) and Tyr aminomutases, (31) and the pyruvoyl group in decarboxylases (Fig. 6) (32). The formation of these cofactors by PTM extends the catalytic power of enzymes greatly, which enables them to catalyze chemistry that is difficult with just the side chains of the 20 amino acids commonly found in proteins.

Figure 5. Fatty acid biosynthesis catalyzed by fatty acid synthases. The growing acyl chain is tethered to the phosphopantetheinylated ACP domain, which enables it to undergo cycles of condensation, ketone reduction, dehydration, and enol reduction catalyzed by different domains. AT, acyltransferase; ACP, acyl-carrier protein; KS, ketosynthase; KR, ketoreductase; DH, dehydratase; ER, enoylreductase.

Figure 6. Post-translationally generated cofactors provide functional groups to allow catalysis. The mechanisms of TOPA quinone in amine oxidases, MIO in deaminases, and pyruvamide in decarboxylases are shown.
Locking proteins into the correct structures or increasing protein stability
The major type of PTM that has this function is protein disulfide bond formation (33). Disulfide bonds are more stable thermodynamically than the reduced thiols in an oxidizing environment. In eukaryotes, proteins that undergo the secretary pathway start to form disulfide bonds once they are translocated into the endoplasmic recticulum (ER) lumen, which is an oxidizing environment. These disulfide bonds help to stabilize the desired protein structure by locking the protein in a certain conformation, and perhaps to assist protein folding too. Many secreted proteins later undergo proteolysis in the Golgi to give smaller fragments (see the proteolysis section below). In this case, disulfide bonds also serve to link the fragments covalently to maintain a certain structure. One textbook example is insulin, which is produced as a single peptide chain that later undergoes several proteolysis step, and the mature insulin consists of two chains connected via two disulfide bonds (Fig. 7) (34). The light and heavy chains of antibodies are connected by disulfide bonds. Another PTM that can increase protein stability is glycosylation. For example, erythropoietin N-glycosylation has been found to increase its in vivo lifetime (35), which is probably because of the blocking of tissue proteases action by carbohydrate modifications.

Figure 7. Maturation of insulin. Insulin is synthesized as preproinsulin that contains an N-terminal signal sequence. After translocating into the ER, the signal sequence is cleaved off by the signal peptidase and the resulting proinsulin folds into a stable conformation. Three disulfide bonds are formed between cysteine side chains. The connecting sequence (Chain C) is cleaved off in the Golgi by proprotein convertases to form the mature and active insulin molecule, which is then secreted.
Exploration of major PTM
In this section, a few major PTM will be explored in more details. For each PTM discussed, a brief introduction on the PTM reaction and the enzymes catalyzing the reaction will be given. A few biological processes that involve the PTM will be explained to demonstrate the important function of the PTM in biology.
Phosphorylation
Protein phosphorylation typically occurs on Ser, Thr, and Tyr residues (Fig. 1), although His and Asp residues can also be phosphorylated as in bacteria two-component signal transduction systems. The universal phosphate donor is adenine triphosphate (ATP, Fig. 8), and the reaction is catalyzed by more than 500 kinases in humans. Many kinases are Ser/Thr specific, some are Tyr specific, whereas some have dual specificity. It was reported that inositol pyrophosphate (IP7) can also serve as phosphate donor in protein phosphorylation (36). However, the reaction is not enzyme catalyzed and the physiologic relevance is not proven yet.
The large number of protein kinases in the human genome reflects that this PTM is widely occurring and regulates numerous biological processes. The most well understood function is signal transduction, because phosphorylation of proteins can turn ON/OFF catalytic activity or create recognition motif to recruit other protein partners, thus allowing signal to propagate. In accord with its role in signal transduction, protein phosphorylation is reversible so that the signaling process can be terminated as needed. The removal of the phosphate group is catalyzed by phosphatases (Fig. 8).

Figure 8. Kinase-catalyzed phosphorylation and phosphatases-catalyzed dephosphorylation reactions. (a) Catalytic mechanism of protein kinases; (b) Catalytic mechanism of bimetallic pSer/pThr or dual specifity protein phosphatases; (c) Catalytic mechanism of pTyr phosphatases.
Two signaling processes will be discussed here to illustrate how protein phosphorylation can play a critical role in cell signaling. A more detailed description of these two signaling processes can be found in the Molecular Cell Biology textbook by Lodish et al. (34). The first one, which is shown in Fig. 9, involves protein kinase A (PKA), which can be activated by cyclic AMP (cAMP) (37). PKA at resting state exists as an inactive tetramer that consists of two copies of a regulatory subunit and two copies of the catalytic subunit. Hormones that signal through G-protein coupled receptors can activate the trimeric G protein, which in turn can activate an effector enzyme, adenylate cyclase (38). Adenylate cyclase catalyzes the formation of cAMP from ATP (39), which results in the increase in cAMP concentration. Binding of cAMP to the regulatory subunits of PKA dissociate the inactive tetramer, which releases the catalytic subunit of PKA. The catalytic subunit can then be activated by phosphorylation at the activation loop. Activated PKA can phosphorylate many different substrates and produce both short-term and long-term effects. Short-term effects come from the change of the catalytic activities of substrate proteins on phosphorylation by PKA. The substrates of PKA include proteins involved in glycogen synthesis and degradation, such as glycogen phosphorylase kinase and glycogen synthase (40). Phosphorylation of these proteins by PKA leads to activation of glycogen degradation and inhibition of glycogen synthesis. Long-term effects come from the changes in gene transcription. PKA can affect transcription by phosphorylating CREB (cAMP response element binding proteins) and other transcription factors (41). On phosphorylation, CREB can bind to specific regions of the chromosomal DNA, and it can recruit the basal transcription machinery via CBP (CREB binding protein)/P300 to activate the transcription of certain genes.

Figure 9. The signaling process that involves G protein-coupled receptors (GPCR) and PKA. (1) Binging of hormone produces conformational change in the GPCR; (2) GPCR binds to Gs protein; (3) GDP bound to Gs is replaced by GTP and the β and γ subunits of Gs dissociate from the α subunit; (4) Gsa subunit binds to adenylate cyclase (AC), which activates the synthesis of cAMP (4a), the hormone tends to dissociate, and hydrolysis of GTP to GDP causes Gsα to dissociate from adenylate cyclase and binds to Gβγ, which regenerates a conformation of Gs that can be activated by an GPCR hormone complex (4b); (5) dissociation of regulatory subunits (R) from PKA as cAMP concentration increases; (6) subsequent activation of the catalytic subunits (C) by phosphorylation in the activation loop generates the fully active kinase; (7) activated PKA can phosphorylate glycogen phosphorylase kinase (GPK) and other enzymes, which leads to activation of glycogen degradation and inhibition of glycogen synthesis; and (8) PKA can affect transcription by phosphorylating the transcription factor CREB.
The second example of cell-signaling process that involves protein phosphorylation is receptor tyrosine kinase signaling (Fig. 10) (42). Receptor tyrosine kinases are transmembrane proteins with an extracellular ligand-binding domain and an intracellular tyrosine kinase domain. Ligand binding to the extracellular domain triggers receptor dimerization and/or activation, so that the intracellular catalytic domains from two receptor protein molecules can phosphorylate each other at the activation segment. This transphosphorylation activates the catalytic domain so that it can phosphorylate other Tyr residues in the receptor and other substrate proteins. These phosphorylated Tyr residues then recruit protein-binding partners that contain SH2 or PTB domains that recognize specific phosphorylated Tyr residues. One of the proteins recruited is Grb2 (growth factor receptor-bound protein 2), which contains an SH2 domain. Grb2 in turn recruit Sos (son of sevenless), which is a guanine nucleotide exchange factor for the G protein Ras. Sos catalyzes the exchange of Ras-bound GDP (guanosine-5’-diphosphate) for GTP (guanosine-5’-tiphosphate), which converts Ras to the activated form. Activated Ras can bind to and activate Raf, which is the most upstream kinase in the MAP kinase (Mitogen-activated protein kinase) cascade (43). By phosphorylation of MEK (MAPK/ERK kinase, a dual specificity MAP kinase kinase) on the activation segment, Raf activates MEK, which in turn phosphorylates and activates ERK. Activated ERK can phosphorylate many transcription factors, which leads to changes in gene transcription and ultimately cell division/differentiation.
The two examples mentioned above illustrated basic principles how protein phosphorylation serves specific biological purposes. Although different kinases might be involved in diverse pathways, the molecular mechanism for the regulation of protein function by phosphorylation is similar: By changing protein structure, phosphorylation can turn ON/OFF the catalytic activity of a protein, or create/mask recognition motif for binding by other molecules.
The 500 or so protein kinases in the human genome regulate numerous biological processes. Consequently, deregulation of protein phosphorylation can lead to various diseases, among which cancer is the most prominent one. Accordingly, kinase inhibitors are being sought for treating various cancers. One best understood example is chronic myeloid leukemia, which is caused by chromosomal abnormality that fuses a kinase ABL (encoded from Abelson gene) with another protein BCR (encoded from breakpoint cluster region gene) (44). The BCR-ABL fusion protein was shown to be sufficient to cause chronic myeloid leukemia in mice. Imatinib mesylate (Gleevec; Novartis Pharmaceuticals, East Hanover, NJ) is a clinically used BCR-ABL inhibitor to treat CML (chronic myelogenous leukemia). The receptor tyrosine kinase and MAP kinase-signaling pathway mentioned above are key pathways that regulate cell proliferation and differentiation; frequently, tumor cells have mutations in proteins involved in this pathway (45). This pathway has thus been studied intensively for the search of cancer drugs. Other kinases, such as cell-division kinases (CDKs), have also been targeted for therapeutics (46). In addition, because phosphatases reverse the effects of kinases, mutations in phosphatases have been indicated in human diseases such as cancer, diabetes, and neurologic disorders (47).

Figure 10. Receptor tyrosine kinase signaling process and the activation of MAP Kinase. (1) Binding of hormone to the receptor causes activation of the kinase activity of the receptor, which leads to phosphorylation of Tyr residues; (2) pTyr residues recruit GRB2, which in turn recruit Sos; (3) Sos promotes exchange of GTP for GDP in Ras, which leads to the active Ras-GTP complex. Then, Sos dissociates from the active Ras; (4) active Ras binds to and activate the kinase Raf (4a) and hormone can dissociate from the receptor (4b); (5) activated Raf phosphoryates and activates MEK; (6) activated MEK phosphorylates and activates of MAP kinase; (7) activated MAP kinase can phosphorylate transcription factors (TF); and (8) phosphorylated translation factors then bind to DNA and lead to changes in gene transcription and ultimately cell division/differentiation.
Acetylation
Acetylation of Lys residues is a very well known PTM because of histone acetylation, which is involved in transcriptional regulation of genes. The acetyl group comes from Acetyl-CoA, and typically, the acetyl acceptor is Lys residues (Fig. 11). Histone acetylation correlates with transcription activation, and accordingly, histone acetyltransferases (HATs) are normally multidomain proteins associated with transcription activator/coactivator complexes (48). The correlation of histone acetylation with transcription activation can be explained by the relaxation of the chromatin structure on histone acetylation and the recruitment of other proteins via acetyl Lys. In eukaryotic cells, chromosomal DNA wrap around core histone octamers consisted of two copies each of histone H2A, H2B, H3 and H4 (49). The complex formed between the histone octamer and the DNA associated with it is called a nucleosome. Nucleosomes can pack into a more condensed structure. Evidence suggests that the tight packing suppresses transcription, whereas transcription activation correlates with relaxed chromatin structure. The N-terminal tails of the histones have many Lys and Arg residues, among other residues, that can be modified post-translationally. No detailed structure information is available to explain how histone tail modification affects nucleosome packing. However, intuitively, masking the positive charges on histones by Lys acetylation can decrease the interaction with negatively charged DNA, which loosens the chromatin structure (50). In addition, acetylated Lys residues can be recognized by proteins that contain bromodomains (Fig. 4) (16, 51), which serve to recruit other proteins (including chromatin remodeling complexes) that help to activate the transcription of the gene.
Histone acetylation not only affects transcription, but also affects other processes that involve DNA, such as nucleosome assembly, heterochromation formation, and DNA repair (52). The acetylation/deacetylation of different Lys residues can have different biological effects. For example, histone H4 Lys5, 8, and 12 acetylation are involved in nucleosome assembly, H4 Lys16 acetylation does not affect nucleosome assembly but is involved in transcription activation (52), whereas H4 Lys56 has been shown recently to promote genome stability and DNA repair in yeast (53, 54).
Proteins other than histones can also be modified by Lys acetylation. Many transcription factors, cytoskeleton proteins, metabolic enzymes, and signaling proteins are acetylated (55). Transcription factors are known to be substrates of HATs, whereas the enzymes responsible for the acetylation of nonnuclear proteins in many cases are not well known (55). The number of proteins that are regulated by acetylation will continue to increase as method to detect protein acetylation improves. Acetylation of nonhistone proteins can change protein-protein interaction, regulate enzymatic activity, and increase protein stability by suppressing ubiquitinylation (55).
Lys acetylation can be reversed by the action of deacetylases. Many deacetylases are Zn-dependent enzymes that use Zn2+ in the active site to activate water molecules to hydrolyze the amide bond (Fig. 11) (56). Recently, another type of deacetylases that are nicotinamide adenine dinucleotide (NAD)-dependent, also known as sirtuins, have been identified (57, 58). Their unique ability to couple NAD degradation to Lys deacetylation (Fig. 11) suggests that this type of enzyme can sense the metabolic state (for example, NAD concentration) of the cell and use that information to regulate the acetylation state and thus the function of the substrate proteins.
In addition to Lys side chain acetylation, protein N-terminal can also be acetylated (59). In eukaryotic cells, the first residue Met in most proteins is cleaved by N-terminal methionine peptidase. The newly released N-terminal amino group is then acetylated. This modification can happen co-translationally before the mature peptide chain is released from the ribosome. The function of this modification in most cases is still not understood, although deletion of the genes involved in this modification has clear phenotypes (59).
Because of the involvement of protein Lys acetylation in regulation of transcription, protein-protein interaction, enzymatic activity, and protein stability, the deregulation of protein acetylation has been associated with many diseases, such as cancer and neurodegeneration (60, 61). Frequently, mutations in histone acetyltransferases are found in cancer (60). Chromosomal abnormalities that generate fusions of acetyltranferases are known to lead to acute myeloid leukemia. These abnormalities include the fusions of MOZ (monocytic leukemia zinc finger protein) with CBP (CREB binding protein) or p300, and fusion of MOZ with the transcription factor TIF2 (transcription intermediary factor 2) (60). MOZ, CBP, p300, and TIF2 all contain histone acetyltransferase domains. Presumably, the generation of these aberrant fusion proteins disrupts normal gene transcription profile, which leads to leukemia. Deregulation of histone deacetylases is also suggested to be associated with cancer (61). A histone deacetylase inhibitor, SAHA (Vorinostat, Merck & Co., Inc, Whitehous Station, NJ), was approved by Food and Drug Administration recently for treatment of cutaneous T-cell lymphoma (62).

Figure 11. (a) Lys acetylation catalyzed by acetyltransferases; (b) mechanism of Zn-dependent HDACs-catalyzed deacetylation; (c) mechanism of sirtuins-catalyzed deacetylation.
Methylation
Although methylation can happen to several different residues (3, 63), most attention has been given to protein Lys/Arg methylation because the methylation of Lys/Arg in histones controls gene transcription. For Lys and Arg methylation, multiple methyl groups can be added to the same Lys or Arg residue (Fig. 12). The methyl group comes from S-adenosyl methionine (SAM), which is a versatile small molecule that is used in many enzymatic transformations (64). Almost all Lys methyltrans- ferases belong to the SET (supressor of variegation-Enhanser of zeste-Trithorax) family of methyltransferases, whereas the protein Arg methyltransferases belong to a different class (65-67). Both histone Lys/Arg methylation and acetylation are associated with transcription regulation. In contrast to histone acetylation, which usually correlates with transcription activation, histone methylation can lead either to transcription activation or to suppression (17, 68). The effect of histone methylation, which is based on current understanding, is mediated by proteins that are recruited by methylated Lys or Arg residues. Tudor domains and chromodomains are known to recognize methylated Lys/Arg residues via both charge interaction and cation-n interaction (69-73). The methylated Lys/Arg residue is more hydrophobic and sterically bulkier than free Lys/Arg, and it can be differentiated by the domains that recognize methylated Lys/Arg residues (69, 74). Sequences that surround the methylate Lys residues are also read by the chromo domains and Tudor domains (69-71). This finding explains why different Lys residues could recruit different proteins on methylation and thus have different biological effects. For example, H3K4 methylation activates transcription by recruiting chromodomain helicase DNA-binding protein 1 (CHD1) specifically in yeast whereas H3K9 methylation represses transcription by recruiting heterochromatin protein 1 (HP1) (75-77).
Nonhistone proteins are known to be methylated on Lys residues, which include transcription factors, such as p53 (78-80), TAF10 (TATA box-binding protein-associated factor 10) (81), and translation factors (63). The p53 protein can be methylated by different methyltransferases [Set9, (78) Smyd2 (79), and Set8 (80)] on different Lys residues (Lys372, 370, and 382, respectively). These different methylation events either activate or repress p53 activity. Arg methylation has been found frequently in nonhistone proteins. For example, PRMT1 has been reported to methylate the transcription factor STAT1 (signal transducers and activators of transcription) (82), PRMT4/CARM1 [coactivator-associated arginine(R) methyltransferase 1] can methylate CBP/p300 (83), and hetergenous nuclear ribonucleoproteins (hnRNPs) and small nuclear ribonucleoproteins (snRNPs) that are involved in pre-mRNA splicing are also Arg methylated (67). The biological functions of these Lys/Arg methylations in most cases can also be explained by the effect of methylation to block or create interaction with other proteins or nucleic acids.
Compared with acetylation, methylation is more stable. For this reason, it was thought that methylation could be a permanent epigenetic mark. The recent discovery of two types of Lys demethylases suggests that methylation is also a reversible PTM. The first Lys demethylase discovered is LSD1 (lysine-specific demethylase 1), which is a FAD (flavin adenine dinucleotide)-dependent enzyme similar to amine oxidases (Fig. 12) (84). It is believed that LSD1 uses two-electron oxidation mechanism and thus cannot demethylate tri-methylated Lys residues (85). The second type of Lys demethylase, which contains the JmjC (Jumonji domain-containing) domain, is a nonheme Fe(II)-dependent enzyme that is capable of doing one-electron oxidation, and thus it can demethylate trimethylated Lys residues (86). The effect of Arg methylation was proposed to be reversed by protein Arg deiminase 4 (PAD4), which generate citrulline via demethyliminiation (87, 88). However, later studies indicate that PAD4 as well as other PAD enzymes do not catalyze demethylimination with appreciable rates in vitro (88-91). A recent report showed that Arg methylation can be truly reversed by JmjC domain containing demethylases, which suggests that PADs are probably not required for Arg demethylation (92). Thus, both Lys and Arg methylation are reversible modifications.
Similar to Lys acetylation, abnormality in Lys methylation has been considered a contributing factor to cancer (93, 94). Decrease in H3 Lys9 and H4 Lys20 trimethyaltion is found in cancer cells. Both H3 Lys9 and H4 Lys20 methylation are associated with heterochromatin formation. Presumably, the decrease in the methyaltion leads to defects in heterochromatin formation, which in turn lead to chromosomal instability and tumor formation (93). Histone methyltransferase fusion proteins generated from chromosomal translocation are found frequently in leukemia and are thought to contribute to the development of leukemia. For example, the H3 Lys79 methyltransferase hDOT1L (human DOT1-like protein) fusion found in mixed lineage leukemia is sufficient to cause leukemic transformation (95). The close association of methylation and cancer suggests that protein methyltranferases and demethylases can be potential therapeutic targets.

Figure 12. (a) Lys/Arg N-methylation; (b) mechanism of FAD-dependent LSDI-catalyzed Lys demethylation; (c) mechanism of Fe-dependent JHDM (JmjC domain-containing histone demethylase)-catalyzed demethylation.
Glycosylation
In eukaryotic cells, glycosylation happens to many membrane and secreted proteins (i.e., proteins that transit through the ER and the Golgi secretary pathway). Glycosylation can occur either on Asn residues (N-glycosylation, Fig. 13), Ser/Thr and post-translationally hydroxylated Lys and Pro residues (O-glycosylation, Fig. 14), or Trp residues (C-glycosylation, Fig. 14). N-glycosylation is a complicated process and involves three stages: 1) the formation of donor substrate with 14 sugar units (GlC3Man9GlcNAc2-PP-dolichol), which occurs in both the cytosolic and the luminal faces of ER (96); 2) the transfer of the tetradecasaccharyl group to the Asn residues found in the consensus sequence Asn-X-Ser/Thr, which occurs in the ER (97); and 3) the hydrolytic removal of the terminal sugar residues on the tetradecasaccharide, the addition of more sugar units (Fig. 13) (98), and the sulfation and phosphorylation of the carbohydrate moieties in the ER and Golgi (99). The later trimming steps can generate different sets of N-linked carbohydrates, such as the high-mannose type glycans, the complex type glycans, and the hybrid type glycans (Fig. 13) (99). Each stage is achieved by the function of multiple proteins. For example, up to nine proteins are required for the transfer of the tetradecasaccharyl group in yeast (100).


Figure 13. Protein N-glycosylation. (1) The formation of the donor substrate with 14 sugar units (Glc3Man9GlcNAc2-PP-dolichol); (2) the reaction scheme that shows the transfer of the tetradecasaccharyl group to the Asn residues found in the consensus sequence Asn-X-Ser/Thr in proteins; (3) hydrolytic removal of the terminal sugar residues on the tetradecasaccharide and addition of more sugar units in the ER and Golgi. OSTase, oligosacchryltransferase.

Figure 14. O- and C-glycosylation reactions. UDP, uridine diphosphate.
Different from N-glycosylation, O-glycosylation starts with the addition of a single sugar residue, which can be followed by the addition of more sugars (101). Similar to N-glycosylation, most O-glycosylation also occurs to proteins that transit through ER and Golgi. However, the addition of a single GlcNAc residue to Ser/Thr is a type of O-glycosylation that occurs to cytosolic proteins (102). This cytosolic O-glycosylation has drawn much attention recently because it can regulate the activity of the substrate proteins, especially because it can compete with protein phosphorylation for the same Ser/Thr on substrate proteins (103).
C-glycosylation is the addition of a single mannosyl group to the indole C-2 position of Trp residues of membrane and secreted proteins (104). The Trp residue that is C-mannosylated reside in a consensus Trp-X-X-Trp sequence, and the first Trp is C-mannosylated. About a dozen proteins in humans are C-mannosylated. The enzyme that catalyzes the modification has not been cloned yet, and currently, the function of this modification is not clear.
The large number of enzymes involved in protein glycosylation and the fact that this complicated N-glycosylation pathway is conserved throughout eukaryotic species suggest that glycosylation has important functions. Deficiency in protein glycosylation causes several diseases in humans, such as lysosomal storage diseases (105), congenital disorders of glycosylation, and leukocytes adhesion deficiency II (106). In addition, changes in glycosylation patterns are associated with cancer and inflammation (107). Protein glycosylation can serve several different biological purposes. One purpose is to help proteins that transit through the secretary pathway to fold correctly. Particularly, the removal of the glucose residue by glucosidase II and the reglucosylation in the ER have been well known to help secreted proteins to fold and make sure only correctly folded proteins are secreted (Fig. 15) (108). Protein O-fucosyltransferase I that modifies Notch protein was reported to have chaperon activity that helps Notch folding and secretion, and this chaperon activity is independent of its catalytic activity (109). Glycosylation is also important for sorting secreted proteins. For example, the phosphorylation of Man on N -glycan (Fig. 16) creates a recognition signal for sorting lysosomal proteins to lysosome. Glycosylation is also believed to increase the protein stability, as has been shown for erythropoietin mentioned earlier. Glycosylation is also proposed to affect ligand receptor interaction and thus regulates cell-cell signaling. However, a detailed molecular understanding about the effect of glycosylation on ligand receptor interaction is hard to obtain in most cases. In two well-studied cases, human CD2 (cellular differentiation marker 2) and IgG (immunoglobulin G), N-glycosylation is found to affect the interaction with their ligands or receptors. Structural data show that the carbohydrate portion does not contact the binding partner directly. Instead, glycsosylation affects the binding by changing the conformation of the glycosylated proteins (110-112).

Figure 15. N-glycosylation helps secreted protein to fold correctly in the ER.
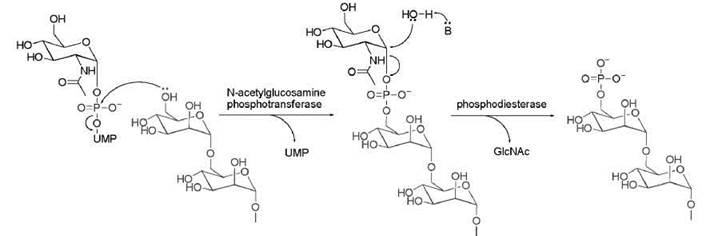
Figure 16. Phophorylation of Man on N-glycan. UMP, uridine monophosphate.
Ubiquitylation
Ubiquitin is an abundant small protein (76 amino acids) found in all eukaryotes. It can be conjugated to many proteins covalently and regulates important biological processes. The addition of ubiquitin to substrate proteins goes through an E1-E2-E3 enzymatic cascade (Fig. 17) (113). E1, which is also called ubiquitin-activating protein (UAP), uses ATP to adenylate the C-terminal Gly of ubiquitin and then captures the activated ubiquitin with a Cys residue in the active site. Most eukaryotic species only have one E1 enzyme responsible for activating all the ubiquitin molecules needed. The ubiquitin-E1 conjugate then is recognized by several dozens of E2 enzymes, which capture ubiquitin from E1 via a transthiolation reaction. The ubiquitin-conjugated E2 enzymes are then recognized by many different E3 enzymes, which recruit the substrate proteins and transfer ubiquitin from E2 to Lys residues of the substrate proteins, either directly or indirectly (Fig. 17). Two major families of E3 enzymes exist: the RING (really interesting new gene) E3s and HECT (homologous to E6AP C terminus) E3s. The Pfam database lists more than 400 RING proteins and 70 HECT proteins. Many E3s form complexes with other proteins. One well-understood E3 complex is the SCF (Skp1-Cullin-F Box) RING E3, for which a crystal structure was reported (114). In humans, multiple Cullins and multiple F Box proteins exist (115). Considering the different combinations, the number of possible E3 complexes can be much more than the number of E3 enzymes (3). E3s decide which substrate proteins get ubiquitylated, thus the large number of E3s and E3 complexes reflects the diverse substrate proteins that must be recognized.
Ubiquitin itself has 7 Lys residues (Lys6, 11, 27, 29, 33, 48, and 63) that can be used for ubiquitin attachment, which lead to polyubiquitylation of substrate proteins. Polyubiquitin chain assembled via different Lys residues have different biological functions (116), as will be explained later. Which Lys residue is used in the polyubiqutine chain is controlled by the specific E3 involved. E3 presumably also controls the length of the polyubiquitin chain, although the detailed chain assembly mechanism is still not clear (117). Ubiquitylation can be reversed by the action of ubiquitin-specific proteases (UBPs). About 60 UBPs exist in the human genome, which presumably recognize different types of ubiquitin modifications at various cellular locations (118).
The biological function of ubiquitylation was recognized originally as targeting proteins to the proteasome for degradation. The importance of this function can be illustrated by many examples. In cell division, progression through the cell cycle is driven by cell division kinases, the activities of which are controlled by a group of proteins called cyclins. Different cyclins function only at certain stages of the cell cycle. Then, they must be degraded, which requires polyubiquitylation by specific E3 enzymes (119). Aberration in the ubiquitylation and degradation of cyclins is associated with cancer. Misfolded proteins must be degraded by the ubiquitin and proteasome system. Aggregation of misfolded proteins is known to cause neurodegeneration, such as Parkinson’s disease (116). Ubiquitylation and proteasome degradation of proteins are also important for other biological processes, such as hypoxia and circadian clock. Ubiquitylation is required for the degradation of hypoxia inducible factor (HIF) on hydroxylation at high oxygen levels (120). Maintaining the circadian clock requires the ubiquitylation and degradation of proteins that inhibit the CLOCK (a protein named from circadian locomotor output cycles kaput gene) transcription factor (121).
It is becoming clear that the biological function of ubiquitylation is not limited to proteasome degradation. Other functions have been discovered, such as promoting membrane protein endocytosis, targeting membrane protein to lysosome for degradation, and regulating cytoplasm/nuclear shuttling (116, 122). It is now generally believed that polyubiquitylation via Lys48 of ubiquitin is a signal for proteasomal degradation, and this action requires minimally 4 ubiquitin units in the chain (123). In contrast, monoubiquitylation, multiple monoubiquitylation on different Lys residues of substrate proteins, and polyubiquitylation via Lys 63 of ubiquitin typically signal proteasome-independent pathways (116). How can so many different functions be achieved? The diverse sets of ubiquitin binding domains (UBDs) provide the molecular explanation to this question (19). Presumably, different UBDs recognize different types of ubiquitin modifications (monoubiquitylation vs. polyubiquitylation, and Lys48-linked vs. Lys63-linked polyubiquitylation, for example), and thus they mediate different functional consequences of ubiquitylation. UBD on yeast proteins Rad23, Rpd10, and Dsk2 recognize the Lys48-linked polyubiquitin chain and deliver the modified substrate proteins to the 26 S proteasome (124). The UBD on the vacuolar proteins recognize monoubiquitylation or Lys63-linked polyubiquitin chain on membrane proteins, which mediate their sorting into lysosome or vacuole. Binding of the Lys63-linked polyubiqutin chain on inhibitor of NF-KB kinase (IKK) by other proteins has been proposed to activate IKK and thus turn on NF-kB signaling (116). The recognition of ubiquitin by UBDs can also explain some “unusual” functions of protein ubiquitylation. For example, Lys48-linked polyubiquitylation of a yeast transcription factor Met4p does not signal for proteasome degradation, but instead it inactivates the transcription factor. It inactivates the transcription factor because Met4p has an in-cis UBD that binds the ubiquitin chain and thus inactivates itself and blocks the proteasomal pathway (125).
In generalization of the function of ubiquitylation, we can say that ubiquitin is an “information-rich protein tag” that can be read by different proteins that contain UBD domains (3), and the exact consequence of ubiquitylation is determined by how the tag is recognized. Besides ubiquitin, eukaryotic cells also have about a dozen known ubiquitin-like protein tags, with SUMO being the best studied one. In addition, many proteins have built-in ubiquitin-like domains. The logic that underlies the biological functions of these ubiquitin-like proteins/domains will likely be the same as what is learned from ubiquitin (3).

Figure 17. Ubiquitylation catalyzed by the E1, E2, E3 cascade.
Proteolysis
Hydrolytic cleavage of proteins by proteases is an irreversible PTM. The large number (more than 500) of proteases in the human genome indicates that proteolysis occurrs often. Proteases can be classified into four types based on catalytic mechanisms (Fig. 18): Ser/Thr proteases, Cys proteases, Asp proteases, and metalloproteases.

Figure 18. Catalytic mechanism of different proteases.
At first glance, proteolysis may seem to be an uncontrolled destruction process like the digestion of food proteins in the gut. In fact, proteolysis in cells is under tight regulation. Even proteases secreted to the digestive tract must be controlled to avoid self-destruction. Typically, proteases are made in the inactive forms (zymogens) that can be activated by proteolysis. Inside eukaryotic cells, two major locations exist for proteolytic degradation of unwanted proteins: the 26 S proteosome and the lysosome (126, 127). Access to the two degradation organelles is controlled tightly. The lysosome is an acidic membrane organelle that contains many proteases and is responsible for degradation of endocytosed membrane proteins, such as activated receptor tyrosine kinases and G protein-coupled receptors that are ubiquitylated and sorted to the lysosome (described in the ubiquitylation section). The lysosome can also degrade endocytosed or phagocytosed bacterial and viral proteins (128). In autophagy, the lysosome is responsible for degrading cellular organelles and some cytosolic protein complexes (126). The 26 S proteosome (Fig. 19) has a 20 S degradation chamber that consists of four rings α β β α (129). In eukaryotes, each a ring has seven different a subunits, and each α ring has seven different β subunits. Three β subunits are catalytically active Thr proteases that are responsible for the degradation of substrate proteins. By forming this chamber, the active sites of the proteases are buried inside the chamber to avoid proteolysis of proteins that should not be digested. Access to the degradation chamber is controlled by the 19 S regulatory complex that caps both ends of the degradation chamber. The regulatory complex contains subunits that recognize polyubiquitylated substrates, subunits that recycle the ubiquitin tag, and subunits that use ATP hydrolysis to unfold and translocate the protein into the degradation chamber. Degradation of the unwanted proteins by the 26 S proteasome or lysosome in a timely fashion is very important. For example, cyclins that activate cell division kinases have to be polyubiquitylated and degraded by the proteasome at specific times to drive cell cycle progression (119). Degradation of activated membrane receptors in the lysosome is important to avoid over stimulation (130, 131). Misfolded proteins must be degraded by the proteasome or lysosome (in autophagy). Failure to do so is thought to contribute to neurodegeneration disorders such as Parkinson’s disease and Alzheimer’s disease (126).

Figure 19. The eukaryotic 26 S proteasome. Subunit compositions of the 19 S regulatory particle of Saccharomyces cerevisiae is shown on the left. The a and p rings of the 20 S proteasome, each of which consists of seven different subunits, are included to indicate how the base 19 S complex is linked to the core 20 S protease complex. The crystal structure of the 20s degradation chamber is shown in both side and top views (figure made using PDB 1RYP).
In addition to the “destructive” proteolysis processes in the proteasome and lysosome, many “constructive” proteolysis processes occur in cells. In both prokaryotes and eukaryotes, secreted proteins contain a signal peptide at the N-terminus that directs them to the secretary pathway. This signal peptide must be cleaved later by signal peptidases (typically serine proteases) so that the protein can transit further in the secretary pathway (132). Many secreted proteins, which include insulin, TGFβ1 (transforming growth factor β1), nerve growth factor β1, albumin, Factor IX, insulin receptor, and Notch, also contain a propeptide that is cleaved by proprotein convertases in the Golgi (133). Selective proteolysis also occurs at the cell membrane in signal transduction processes. Notch protein, on binding to its ligand Delta/Jagged (membrane proteins on neighboring cells), is cleaved by one of the ADAM (a disintegrase and metallo-protease) proteins at a site close to the transmembrane region. This cleavage activates Notch for regulated intramembrane proteolysis, which cuts within the membrane-spanning region of Notch and releases the intracellular domain of Notch from the cytoplasm membrane. Then, the intracellular domain translocates into the nucleus where it acts as a transcription factor to turn on genes required for development (Fig. 20) (134). Regulated intramembrane proteolysis is catalyzed by the membrane protein complex called presenilin that contains Asp protease subunits. Presenilin is also responsible for cleavage of the amyloid-p precursor proteins in Alzheimer’s disease. This proteolysis-triggered proteolysis signaling occurs often. Similar signaling pathways are present also in bacteria. For example, the release of the transcription factor σEis achieved via the sequential cleavage of the membrane protein RseA by DegS (a Ser protease) and YaeL (a Zn protease) (135).

Figure 20. Four proteolysis events for Notch that lead to the release of an active transcription factor. TGN, trans Golgi network.

Figure 21. (a) Domain structures of mammalian caspases; (b) the caspase cascades and the initiation of apoptosis. Apaf-1, apoptotic protease activation factor-1; Cyto c, cytochrome c; FADD, Fas-associated protein with death domain; LS, large subunit; RAIDD, RIP-associated ICH-1/CED-3 homologous protein with a death domain; RIP, receptor-interacting protein; TRADD, tumor necrosis factor receptor-associated protein with death domain; SS, small subunit.
Similar to the MAP kinase cascades for protein phosphorylation, protease cascades exist, in which downstream proteases are activated by the action of upstream proteases (3). One of the most famous cascades is the caspase cascade that leads to apoptosis (Fig. 22) (11, 136). Caspases are Cys proteases that cleave the amide bond specifically after an Asp residue. Two types of caspases exist, initiator caspases (Caspase 2, 8, 9, 10) and effector caspases (Caspase 3, 6, 7). Both initiator and effector caspases are produced in zymogen forms. Initiator caspases use their N-terminal DED (death effector domain) and CARD (caspase recruitment domain) domains to interact with other proteins to receive apoptosis signals. The signals cause the dimerization of the initiator caspases and activate them so that they can cleave themselves and the effector caspases after specific Asp residues. Cleavage by the initiator caspases activates the effector caspases, which then cleave their substrate proteins to carry out cell apoptosis. The substrate proteins of effector caspases include the inhibitor of caspases-activated DNAse (deoxyribonuclease), Bcl2 (named from B-cell lymphoma 2, an antiapoptotic protein), and PARP-1 (poly(ADP-ribose) polymerase-1, an enzyme catalyzing protein poly(ADP-ribosyl)ation and required for DNA repair). Cleavage of the inhibitor of DNAse by effector caspases activates its catalytic activity, resulting in the fragmentation of chromosomal DNA, which is a hallmark of apoptosis. The caspases cascade and apoptosis is very important for the development and homeostasis of metazoans. Decreased ability of cells to undergo apoptosis will lead to cancer, whereas too much apoptosis can lead to autoimmune diseases (137).

Figure 22. Shokat's (144) ''bump and hole'' method to identify substrates for kinases.
Identifying new pathways regulated by known PTM and discovering new PTM
The brief description above on a few major PTM demonstrates clearly that PTM can regulate many important biological processes. So far, a fairly good understanding of many aspects of PTM has been obtained. What remaining challenges must be addressed?
One direction is to figure out the molecular details of many of the biological processes that are regulated by PTM. Structural biology and biochemisty is needed to answer questions like what structural changes are induced by a particular PTM and how the structure changes lead to changes in activity or recognition by binding partners. Much progress has been made in this direction but still more remains to be figured out. For example, in protein ubiquitylation, no structural details about E1 exist, it is not clear how the polyubiquitin chain is made (117), and it is not clear how specificities of different ubiquitin binding domains are achieved (19).
Another direction is to identify the proteome that is modified by a specific PTM. Advancement in protein identification by mass spectrometry (MS) has greatly facilitated studies in this direction and many efforts have been invested. Generally, an affinity purification method is used to enrich proteins that are modified by a specific PTM, and then these proteins are identified by MS. For example, phosphotyrosine-specific antibodies have been used to enrich proteins that are modified on Tyr residues, and metal affinity columns have been used to isolate all phosphopeptides (138). These isolated phosphoproteins/peptides can then be identified by MS. A His6 tag has been fused to the N-terminus of ubiquitin and used to isolate ubiquitylated proteins that are then identified by MS (139). GlcNAc with an azide group attached has been used to label proteins that are O-GlcNAc modified, and then a biotin tag is conjugated to the modified protein via Staudinger ligation. O-GlcNAc modified proteins can be pulled out using streptavidin beads and identified using MS. Using this method, close to 200 O-GlcNAc modified proteins were identified (140). A clever method to detect protein S-acylation has been reported recently (141).
These proteomic studies have provided much information. However, to understand the function of a PTM in cell physiology completely, it is desirable to know which enzyme is responsible for the modification of a particular substrate protein. With the availability of bioinformatics tools and completed genome sequences, it is now relatively straightforward to identify all the enzymes in a genome that share similar biochemical function. For example, we now know that the human genome contains more than 500 protein kinases, more than 500 proteases, and ~400 ubiquitin E3s. But without knowing what substrate proteins they modify, it will be very difficult (if not impossible) to understand their biological functions on a molecular level. Currently, no efficient and reliable method exists yet to identify the substrate proteins for an enzyme. A straightforward method is to make a library of short peptides and try to identify consensus sequences that are recognized by an enzyme (142, 143). The disadvantage is that the structure of a short peptide may be different from the structure of the same sequence present in a folded protein. Thus, the reliability of this method must be validated by other methods. Shokat and coworkers (144) have used a clever approach to identify kinase substrates (Fig. 22). This approach uses a bulky ATP analog that can be used only by a kinase mutant as a cosubstrate. By incubating 32P-labeled ATP analog and the kinase mutant with cell extract, the substrate proteins of the specific kinase can be labeled. Identification of the substrate proteins may be difficult though because the radiolabeled substrate proteins cannot be enriched/purified easily for identification by MS. It is not clear whether this method can be applied easily to other PTM enzymes.
Parallel to the efforts of identifying substrate proteins for a particular enzyme, the activity-based small molecule probes pioneered by Cravatt and coworkers can facilitate the identification of the biological functions of an enzyme that catalyzes protein post-translational modifications (145). The major advantage of this type of probes is that potentially they can detect enzymes that are in the active states, and thus can provide snapshots of enzymes that are in the active states at different development stages or different types of cells. Among enzymes that catalyze PTM, so far probes have been developed for studying proteases (145, 146), kinases (147), pTyr phosphatases (148), and protein Arg deiminases (149).
Perhaps a more challenging question is how we can discover new PTM reactions. In principle, there are analytic tools that can be used to research this topic. One such tool is top-down FT-MS, which determines the molecular weight of the whole protein with high accuracy. By comparing the obtained tandem MS (MS/MS) result with the expected MS/MS result, post-translational modifications can be identified (150). Crystallography can also discover new PTM, if a protein expressed in the proper host can be crystallized. Some rare modifications or protein side chains were discovered this way (151). However, the success of using these methods would require that a significant portion of the protein population is modified and the modification is stable. This condition cannot be met by all PTM. Thus, discovering new PTM poses a great challenge to chemical biologists. Undoubtedly, new PTM reactions are waiting to be discovered and the identification of these new PTM, together with the identification of new pathways that are regulated by known PTM, will advance our understanding about the molecular logic of living systems.
References
1. International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001; 409:860-921.
2. Puente XS, Sanchez LM, Overall CM, Lopez-Otin C. Human and mouse proteases: a comparative genomic approach. Nat. Rev. Genet. 2003; 4:544-558.
3. Walsh CT. Posttranslational Modification of Proteins: Expanding Nature’s Inventory. 2005. Roberts and Company Publishers, Englewood, CO.
4. Ahmed N, Thornalley PJ. Advanced glycation endproducts: what is their relevance to diabetic complications? Diabetes Obes. Metab. 2007; 9:233-245.
5. Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 2005; 6:150-166.
6. Lippard SJ, Berg JM. Principles of Bioinorganic Chemistry. 1994. University Science Books. Mill Valley, CA.
7. Ruthenburg AJ, et al. Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex. Nat. Struct. Mol. Biol. 2006; 13:704-712.
8. Johnson LN, Lewis RJ. Structural basis for control by phosphorylation. Chem. Rev. 2001; 101:2209-2242.
9. Johnson LN, Barford D. The effects of phosphorylation on the structure and function of proteins. Annu. Rev. Biophys. Biomol. Struct. 1993; 22:199-232.
10. Chen P, Hochstrasser M. Autocatalytic subunit processing couples active site formation in the 20S proteasome to completion of assembly. Cell 1996; 86:961-972.
11. Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004; 5:897-907.
12. Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science 2003; 300:445-452.
13. Yaffe MB, Elia, AEH. Phosphoserine/threonine-binding domains. Curr. Opin. Cell Biol. 2001; 13:131-138.
14. Yaffe MB. Phosphotyrosine-binding domains in signal transduction. Nat. Rev. Mol. Cell Biol. 2002; 3:177-186.
15. Xiang-Jiao Y. Lysine acetylation and the bromodomain: a new partnership for signaling. Bioessays 2004; 26:1076-1087.
16. Mujtaba S, Zeng L, Zhou MM. Structure and acetyl-lysine recognition of the bromodomain. Oncogene 2007; 26:5521-5527.
17. Daniel JA, Pray-Grant MG, Grant PA. Effector proteins for methylated histones. Cell Cycle 2005; 4:919-926.
18. Hurley JH, Lee S, Prag G. Ubiquitin-binding domains. Biochem. J. 2006; 399:361-372.
19. Harper JW, Schulman BA. Structural complexity in ubiquitin recognition. Cell 2006; 124;1133-1136.
20. Pittet M, Conzelmann A. Biosynthesis and function of GPI proteins in the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 2007; 1771:405-420.
21. Farazi TA, Waksman G, Gordon JI. The biology and enzymology of protein N-myristoylation. J. Biol. Chem. 2001; 276:39501-39504.
22. McTaggart S. Isoprenylated proteins. Cell. Mol. Life Sci. 2006; 63:255-267.
23. Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 2007; 8:74-84.
24. Resh MD. Trafficking and signaling by fatty-acylated and prenylated proteins. Nat. Chem. Biol. 2006; 2:584-590.
25. Perham RN. Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annu. Rev. Biochem. 2000; 69:961-1004.
26. Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem. Rev. 2006; 106:3468-3496.
27. Schwartz B, Klinman JP. Mechanisms of biosynthesis of protein-derived redox cofactors. Vitam. Horm. 2001; 61:219-239.
28. Ghosh D. Human sulfatases: a structural perspective to catalysis. Cell. Mol. Life Sci. 2007; 64:2013-2022.
29. Schwede TF, Retey J, Schulz GE. Crystal structure of histidine ammonia-lyase revealing a novel polypeptide modification as the catalytic electrophile. Biochemistry 1999; 38:5355-5361.
30. Calabrese JC, Jordan DB, Boodhoo A, Sariaslani S, Vannelli T. Crystal structure of phenylalanine ammonia lyase: multiple helix dipoles implicated in catalysis. Biochemistry 2004; 43:11403-11416.
31. Christenson SD, Liu W, Toney, MD, Shen B. A novel 4-methylideneimidazole-5-one-containing tyrosine aminomutase in enediyne antitumor antibiotic C-1027 biosynthesis. J. Am. Chem. Soc. 2003; 125:6062-6063.
32. Poelje PD, Snell EE. Pyruvoyl-dependent enzymes. Annu. Rev. Biochem. 1990; 59:29-59.
33. Kadokura H, Katzen F, Beckwith J. Protein disulfide bond formation in prokaryotes. Annu. Rev. Biochem. 2003; 72:111-135.
34. Lodish H, et al. Molecular Cell Biology. 2007. W.H. Freeman & Co Ltd, New York.
35. Takeuchi M, Kobata A. Structures and functional roles of the sugar chains of human erythropoietins. Glycobiology 1991; 1:337-346.
36. Saiardi A, Bhandari R, Resnick AC, Snowman AM, Snyder SH. Phosphorylation of proteins by inositol pyrophosphates. Science 2004; 306:2101-2105.
37. Skalhegg BS, Tasken K. Specificity in the cAMP/PKA signaling pathway. Differential expression,regulation, and subcellular localization of subunits of PKA. Front. Biosci. 2000; 5:678-693.
38. Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002; 3:639-650.
39. Hurley JH. Structure, mechanism, and regulation of mammalian adenylyl cyclase. J. Biol. Chem. 1999; 274:7599-7602.
40. Krebs EG, Beavo JA. Phosphorylation-dephosphorylation of enzymes. Annu. Rev. Biochem. 1979; 48:923-959.
41. Daniel PB, Walker WH, Habener JF. Cyclic amp signaling and gene regulation. Ann. Rev. Nutr. 1998; 18:353-383.
42. Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2000; 103:211-225.
43. Avruch J. MAP kinase pathways: the first twenty years. Biochim. Biophys. Acta 2007; 1773:1150-1160.
44. Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Oncogene 2007; 7:441-453.
45. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen- activated protein kinase cascade for the treatment of cancer. Oncogene 2007; 26:3291-3310.
46. Shchemelinin I, Sefc L, Necas E. Protein kinase inhibitors. Folia Biol. (Praha) 2006; 52:137-148.
47. Laurent Bialy HW. Inhibitors of protein tyrosine phosphatases: next-generation drugs? Angew. Chem. Int. Ed. Engl. 2005; 44:3814-3839.
48. Roth SY, Denu JM, Allis CD. Histone acetyl transferases. Annu. Rev. Biochem. 2001; 70:81-120.
49. Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997; 389:251-260.
50. Verdone L, Caserta M, Mauro ED. Role of histone acetylation in the control of gene expression. Biochem. Cell Biol. 2005; 83:344-353.
51. Yang XJ. Lysine acetylation and the bromodomain: a new partnership for signaling. Bioessays 2004; 26:1076-1087.
52. Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007; 76:75-100.
53. Han J, et al. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 2007; 15:653-655.
54. Driscoll R, Hudson A, Jackson SP. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science 2007; 315:649-652.
55. Yang XJ, Gregoire S. Metabolism, cytoskeleton and cellular signalling in the grip of protein Ne - and O-acetylation. EMBO Rep. 2007; 8:556-562.
56. Grozinger CM, Schreiber SL. Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chem. Biol. 2002; 9:3-16.
57. Imai SI, Armstrong CM, Kaeberlein M, Guarente L Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000; 403:795-800.
58. Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu. Rev. Biochem. 2006; 75:435-465.
59. Polevoda B, Sherman F. N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J. Mol. Biol. 2003; 325:595-622.
60. Timmermann S, Lehrmann H, Polesskaya A, Harel-Bellan A. Histone acetylation and disease. Cell. Mol. Life Sci. 2001; 58:728-736.
61. Varier RA, Swaminathan V, Balasubramanyam K, Kundu TK. Implications of small molecule activators and inhibitors of histone acetyltransferases in chromatin therapy. Biochem. Pharmacol. 2004; 68:1215-1220.
62. Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007; 25:84-90.
63. Polevoda B, Sherman F. Methylation of proteins involved in translation. Mol. Microbiol. 2007; 65:590-606.
64. Fontecave M, Atta M, Mulliez E. S-adenosylmethionine: nothing goes to waste. Trends Biochem. Sci. 2004; 29:243-249.
65. Kouzarides T. Histone methylation in transcriptional control. Curr. Opin. Genet. Dev. 2002; 12:198-209.
66. Schubert HL, Blumenthal RM, Cheng X. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 2003; 28:329-335.
67. Bedford MT, Richard S. Arginine methylation: an emerging regulator of protein function. Mol. Cell 18, 263-272 (2005).
68. Bannister AJ, Kouzarides T. Reversing histone methylation. Nature 2005; 436:1103-1106.
69. Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science 2002; 295:2080-2083.
70. Huyen Y, et al. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 2004; 432:406-411.
71. Flanagan JF, et al. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature 2005; 438:1181-1185.
72. Cote J, Richard S. Tudor domains bind symmetrical dimethylated arginines. J. Biol. Chem. 2005; 280:28476-28483.
73. Sprangers R, Groves MR, Sinning I, Sattler M. High-resolution X-ray and NMR structures of the SMN tudor domain: conformational variation in the binding site for symmetrically dimethylated arginine residues. J. Mol. Biol. 2003; 327:507-520.
74. Friesen WJ, Massenet S, Paushkin S, Wyce A, Dreyfuss G. SMN, the product of the spinal muscular atrophy gene, binds preferentially to dimethylarginine-containing protein targets. Mol. Cell 2001; 7:1111-1117.
75. Bannister AJ, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001; 410:120-124.
76. Nakayama J-I, Rice JC, Strahl BD, Allis CD, Grewal SIS. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 2001; 292:110-113.
77. Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001; 410:116-120.
78. Chuikov S, et al. Regulation of p53 activity through lysine methylation. Nature 2004; 432:353-360.
79. Huang J, et al. Repression of p53 activity by Smyd2-mediated methylation. Nature 2006; 444:629-632.
80. Shi X, et al. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 2007; 27:636-646.
81. Kouskouti A, Scheer E, Staub A, Tora L, Talianidis I. Genespecific modulation of TAF10 function by SET9-mediated methylation. Mol. Cell 2004; 14:175-182.
82. Mowen KA, et al. Arginine methylation of STAT1 modulates IFN[alpha]/[beta]-induced transcription. Cell 2001; 104:731-741.
83. Xu W, et al. A transcriptional switch mediated by cofactor methylation. Science 2001; 294:2507-2511.
84. Shi Y, et al. Histone demethylation mediated by the nuclear amine oxidase Homolog LSD1. Cell 2004; 119:941-953.
85. Stavropoulos P, Blobel G, Hoelz A. Crystal structure and mechanism of human lysine-specific demethylase-1. Nat. Struct. Mol. Biol. 2006; 13:626-632.
86. Whetstine JR, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 2006; 125:467-481.
87. Wang Y, et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 2004; 306:279-283.
88. Thompson PR, Fast W. Histone citrullination by protein arginine deiminase: is arginine methylation a green light or a roadblock? ACS Chem. Biol. 2006; 1:433-441.
89. Kearney PL, et al. Kinetic characterization of protein arginine deiminase 4: a transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry 2005; 44:10570-10582.
90. Raijmakers R, et al. Methylation of arginine residues interferes with citrullination by peptidylarginine deiminases in vitro. J. Mol. Biol. 2007; 367:1118-1129.
91. Hidaka Y, Hagiwara T, Yamada M. Methylation of the guanidino group of arginine residues prevents citrullination by peptidylarginine deiminase IV. FEBS Lett. 2005; 579:4088-4092.
92. Chang B, Chen Y, Zhao Y, Bruick RK. JMJD6 is a histone arginine demethylase. Science 2007; 318:444-447.
93. Fraga MF, Esteller M. Towards the human cancer epigenome: a first draft of histone modifications. Cell Cycle 2005; 4:1377-1381.
94. Shi Y, Whetstine Jr. Dynamic regulation of histone lysine methylation by demethylases. Mol. Cell 2007; 25:1-14.
95. Okada, Y. et al. hDOT1L links histone methylation to leukemogenesis. Cell 2005; 121:167-178.
96. Burda P, Aebi M. The dolichol pathway of N-linked glycosylation. Biochim. Biophys. Acta 1999; 1426:239-257.
97. Yan A, Lennarz WJ. Unraveling the mechanism of protein N-glycosylation. J. Biol. Chem. 2005; 280:3121-3124.
98. Roth J. Protein N-glycosylation along the secretory pathway: relationship to organelle topography and function, protein quality control, and cell interactions. Chem. Rev. 2002; 102:285-304.
99. Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 1985; 54:631-664.
100. Knauer R, Lehle L. The oligosaccharyltransferase complex from yeast. Biochim. Biophys. Acta 1999; 1426:259-273.
101. PeterKatalinic J. Methods in enzymology: O-Glycosylation of proteins. Methods Enzymol. 2005; 405:139-171.
102. Zachara NE, Hart GW. The emerging significance of O-GlcNAc in cellular regulation. Chem. Rev. 2002; 102:431-438.
103. Love DC, Hanover JA. The hexosamine signaling pathway: deciphering the “O-GlcNAc Code”. Sci. STKE 20051;re13.
104. Furmanek A, Hofsteenge J. Protein C-mannosylation: facts and questions. Acta Biochim Pol. 2000; 47:781-789.
105. Neufeld EF. Lysosomal storage diseases. Annu. Rev. Biochem. 1991; 60:257-280.
106. Schachter H. Congenital disorders involving defective N-glycosylation of proteins. Cell. Mol. Life Sci. 2001; 58:1085-1104.
107. Dube DH, Bertozzi CR. Glycans in cancer and inflammation - potential for therapeutics and diagnostics. Nat. Rev. Drug Discov. 2005; 4:477-488.
108. Trombetta ES, Parodi AJ. Quality control and protein folding in the secretory pathway. Annu. Rev. Cel. Dev. Biol. 2003; 19:649-676.
109. Okajima T, Xu A, Lei L, Irvine KD. Chaperone activity of protein O-fucosyltransferase 1 promotes notch receptor folding. Science 2005; 307:1599-1603.
110. Wyss DF, et al. Conformation and function of the N-linked glycan in the adhesion domain of human CD2. Science 1995; 269:1273-1278.
111. Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J. Mol. Biol. 2003; 325:979-989.
112. Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment-FcyRIII complex. Nature 2000; 406:267-273.
113. Pickart CM. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001; 70:503-533.
114. Zheng N, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 2002; 416:703-709.
115. Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 2004; 5:739-751.
116. Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007; 315:201-205.
117. Hochstrasser M. Lingering mysteries of ubiquitin-chain assembly. Cell 2006; 124:27-34.
118. Wing SS. Deubiquitinating enzymes-the importance of driving in reverse along the ubiquitin-proteasome pathway. Int. J. Biochem. Cell Biol. 2003; 35:590-605.
119. Reed SI. Ratchets and clocks: the cell cycle, ubiquitylation and protein turnover. Nat. Rev. Mol. Cell Biol. 2003; 4:855-864.
120. Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004; 5:343-354.
121. Gallego M, Virshup DM. Post-translational modifications regulate the ticking of the circadian clock. Nat. Rev. Mol. Cell Biol. 2007; 8:139-148.
122. Salmena L, Pandolfi PP. Changing venues for tumour suppression: balancing destruction and localization by monoubiquitylation. Nat. Rev. Cancer 2007; 7:409-413.
123. Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000; 19:94-102.
124. Madura K. Rad23 and Rpn10: perennial wallflowers join the melee. Trends Biochem. Sci. 2004; 29:637-640.
125. Flick K, Raasi S, Zhang H, Yen JL, Kaiser P. A ubiquitin-interacting motif protects polyubiquitinated Met4 from degradation by the 26S proteasome. Nat. Cell Biol. 2006; 8:509-515.
126. Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006; 443:780-786.
127. Aaron C. Intracellular protein degradation: from a vague idea, through the lysosome and the ubiquitin-proteasome system, and onto human diseases and drug targeting (nobel lecture). Angew. Chem. Int. Ed. 2005; 44:5944-5967.
128. Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat. Rev. Mol. Cell Biol. 2007: 8:622-632.
129. Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 1999; 68:1015-1068.
130. Marmor MD, Yarden Y. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene 2004; 23:2057-2070.
131. Shenoy SK. Seven-transmembrane receptors and ubiquitination. Circ. Res. 2007; 100:1142-1154.
132. Paetzel M, Karla A, Strynadka NCJ, Dalbey RE. Signal peptidases. Chem. Rev. 2002; 102:4549-4580.
133. Rockwell NC, Krysan DJ, Komiyama T, Fuller RS. Precursor processing by Kex2/Furin proteases. Chem. Rev. 2002; 102:4525-4548.
134. Fortini ME. [gamma]-Secretase-mediated proteolysis in cell-surface-receptor signalling. Nat. Rev. Mol. Cell Biol. 2002; 3:673-684.
135. Young JC, Hartl FU. A stress sensor for the bacterial periplasm. Cell 2003; 113:1-2.
136. Yan N, Shi Y. Mechanisms of apoptosis through structural biology. Annu. Rev. Cell. Dev. Biol. 2005; 21:35-56.
137. Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science 1995; 267:1456-1462.
138. Kalume DE, Molina H, Pandey A. Tackling the phosphoproteome: tools and strategies. Curr. Opin. Chem. Biol. 2003; 7:64-69.
139. Peng J, et al. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 2003; 21:921-926.
140. Nandi A, et al. Global identification of O-GlcNAc-modified proteins. Anal. Chem. 2006; 78:452-458.
141. Roth AF, et al. Global analysis of protein palmitoylation in yeast. Cell Cycle 2006; 125:1003-1013.
142. Songyang Z, et al. Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr. Biol. 1994; 4:973-982.
143. Obata T, et al. Peptide and protein library screening defines optimal substrate motifs for AKT/PKB. J. Biol. Chem. 2000; 275:36108-36115.
144. Ubersax JA, et al. Targets of the cyclin-dependent kinase Cdk1. Nature 2003; 425:859-864.
145. Evans MJ, Cravatt BF. Mechanism-based profiling of enzyme families. Chem. Rev. 2006; 106:3279-3301.
146. Love KR, Catic A, Schlieker C, Ploegh HL. Mechanisms, biology and inhibitors of deubiquitinating enzymes. Nat. Chem. Biol. 2007; 3:697-705.
147. Patricelli MP, et al. Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry 2007; 46:350-358.
148. Kumar S, et al. Activity-based probes for protein tyrosine phosphatases. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:7943-7948.
149. Luo Y, Knuckley B, Bhatia M, Pellechia PJ, Thompson PR. Activity-based protein profiling reagents for protein arginine deiminase 4 (PAD4): synthesis and in vitro evaluation of a fluorescently labeled probe. J. Am. Chem. Soc. 2006; 128:14468-14469.
150. Sze SK, Ge Y, Oh H, McLafferty FW. From the cover: Top-down mass spectrometry of a 29-kDa protein for characterization of any posttranslational modification to within one residue. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:1774-1779.
151. Ermler U, Grabarse W, Shima S, Goubeaud M, Thauer RK. Crystal structure of methyl-coenzyme M reductase: the key enzyme of biologic methane formation. Science 1997; 278:1457-1462.