CHEMICAL BIOLOGY
Protease Inhibitors, Mechanisms of
Christopher J. Farady, University of California, San Francisco Charles S. Craik, University of California, San Francisco
doi: 10.1002/9780470048672.wecb474
Relatively few design principles underlie the mechanisms of inhibition of a myriad range of protease inhibitors. Protease inhibitors tend to be competitive and to compete with substrate binding, either through direct competition or deformation of the protease active site. Although protein inhibitors can gain potency through the burial of a large surface area and specificity through contacts with specific exosites, small-molecule inhibitors primarily gain potency through interactions with the catalytic machinery of the enzyme and specificity through interactions with the substrate binding sites. Incorporation of these design principles into chemical probes and drugs have improved greatly our ability to create potent and specific protease inhibitors.
Proteolytic enzymes are ubiquitous in all organisms and constitute 2-4% of the encoded gene products. They are critical for diverse biologic processes such as digestion, blood clotting, host defense, pathogenic infection, viral replication, wound healing, and disease progression, to name a few. Because proteases trigger an irreversible event—the cleavage of a protein—their activity must be controlled tightly. Dysregulated proteolytic activity causes a disruption in the homeostatic balance of a biologic system and can result in any number of poor biologic outcomes. As a result, nature has developed several strategies for inhibiting proteases to control proteolysis. Similar approaches have often been employed in the development of synthetic protease inhibitors. To a large extent, the same design principles that work well for naturally occurring protease inhibitors work well for inhibitors developed in the laboratory.
This review aims to survey the mechanisms by which protease inhibitors function. To achieve this goal, we have divided inhibitors into categories based on their mechanism, to illustrate that a relatively small number of design principles can be combined to develop new and effective protease inhibitors. These divisions are somewhat arbitrary, as many inhibitors could be grouped into several classes. Because of space limitations, the list of mechanisms is not exhaustive in its treatment of all inhibitors, but it aims to be illustrative of the many ways proteases can be inhibited. Although beyond the scope of this review, it is also important to keep in mind that spatial and temporal regulation of proteolytic activity is critically important in biology. Beyond the many levels of transcriptional and translational control, proteases are expressed as inactive (or nearly inactive) zymogens, and they are not activated until needed. Furthermore, they are often localized to cellular structures such as the cell membrane or stored in specific organelles such as lysozomes or granules to minimize unwanted proteolysis. Background on the four major classes of proteases (serine, cysteine, aspartic, and metalloproteases) as well as on the basic mechanisms of enzyme inhibition is abundant; please consult the “Further Reading” list at the end of the chapter. Figure 1 provides an overview of basic substrate and protease nomenclature, whereas Tables 1 and 2 list many of the inhibitors discussed in the text (1-21).
Table 1. Naturally occurring protein protease inhibitors
|
Inhibitor |
Target protease |
Mechanism |
Specificity |
KI |
Reference |
|
BPTI |
S1 serine proteases |
Standard Mechanism |
broad specificity |
femtomolar to low nanomolar |
(1) |
|
SMPI |
M4 metalloproteases |
Standard? |
fold-specific |
thermolysin -0.1 nM |
(3) |
|
Staphostatin B |
staphopain B |
Competitive |
specific |
<5 nM |
(4) |
|
Cystatin A |
papain family cysteine proteases |
Competitive |
broad specificity |
picomolar to low nanomolar |
(19) |
|
TIMP 1 |
matrix metalloproteases |
Competitive |
broad specificity |
low nanomolar |
(6) |
|
Ascaris pepsin inhibitor 3 |
aspartic proteases, cathepsin E, pepsin, gastracin |
Competitive |
some specificity |
1-100 nM |
(7) |
|
Ecotin |
S1 serine proteases |
Competitive, Exosite binding |
fold-specifc |
picomolar to low nanomolar |
(44) |
|
Hirudin |
thrombin |
Competitive, Exosite binding |
specific |
0.2 pM |
(11) |
|
XIAP-BIR3 |
caspase-9 |
Allosteric, Competitive |
specific |
20 nM |
(14) |
|
α-2-macroglobin |
most proteases |
Activity dependent |
non-specific |
N.D. |
(18) |
|
α-1-antipeptidase (serpin) |
serine, occasionally cysteine |
Activity dependent |
broad specificity |
elastase-6.5 x 107 M-1s-1 |
(17) |
Table 2. Small-molecule protease inhibitors
|
Inhibitor |
Target protease |
Mechanism |
Specificity |
KI1 |
Reference |
|
Bortezomib |
proteosome |
competitive, transition state analog |
specific |
proteosome 0.62 nM chymotrypsin 320 nM |
(43) |
|
Pepstatin A |
aspartic proteases |
competitive, transition state analog |
class-specific |
sub-nanomolar |
(18) |
|
Idinavir |
HIV protease |
competitive, transition state analog |
specific |
HIV1 pr- 0.35 nM c. albicans asp protease- 1 μM |
(42) |
|
Leupeptin |
serine, cysteine, threonine proteases |
competitive, transition state analog |
P1 arginine specificity |
trypsin 130 nM cathepsin B- 6 nM |
(51) |
|
E64 |
cysteine proteases |
irreversible, alkylation |
C1 class-specific |
cathepsin B 89,400 M-1s-1 |
(23) |
|
Phosphonates* |
serine proteases |
irreversible, phosphonylation |
P1 residue specificity |
thrombin 700 M-1s-1 trypsin 110 M-1s-1 |
(47) |
|
Cephalosporin analog |
elastase |
mechanism-based |
some cross-reactivity |
161,000 M-1s-1 |
(48) |
|
Melagatran |
thrombin |
competitive, active site |
some cross-reactivity |
thrombin - 2 nM trypsin 3.6 nM |
(49) |
|
Marimastat |
matrix metalloproteases |
competitive, active site |
inhibits many MMPs |
inhibits MMP 1,2,7,8,9 ICt0 < 20 nM |
(50) |
|
Captopril |
angiotensin converting enzyme |
competitive, active site |
specific |
ACE- 23 nM (IC50) aminopeptidaseP- 110 μM |
(45), (46) |
1 -Inhibition constants for covalent inhibitors are given as kinact/KI.
* Values are for D-Phe-Pro-(4AmPhGly)P(OPh)2

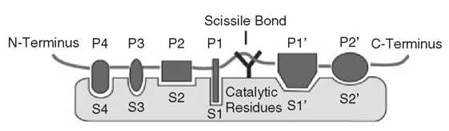
Figure 1. Diagram of a protease active site. A protease cleaves a peptide at the scissile bond, and has a number of specificity subsites, which determine protease specificity. Substrates bind to a protease with their non-prime residues on the N-terminal side of the scissile bond and their prime-side residues C-terminal to the scissile bond. The catalytic residues determine the class of protease. Serine, cysteine, and threonine proteases hydrolyze a peptide bond via a covalent acyl-enzyme intermediate, and aspartic, glutamic and metalloproteases activate a water molecule to hydrolyze the peptide bond in a non-covalent manner.
Mechanisms of Naturally Occurring Protein Protease Inhibitors
Standard mechanism
The most thoroughly studied mechanism of protein protease inhibitors is that of the standard mechanism (or Canonical or Laskowski mechanism) inhibitors of serine proteases (1) (Fig. 2). Standard mechanism inhibitors include the Kazal, Kunitz, and Bowman-Birk family of inhibitors and bind in a lock-and-key fashion. All standard mechanism inhibitors insert a reactive loop into the active site of the protease, which is complementary to the substrate specificity of the target protease and binds in an extended fi-sheet with the enzyme in a substrate-like manner. While bound to the protease, the “scissile bond” of standard mechanism inhibitors is hydrolyzed very slowly, but products are not released and the amide bond is re-ligated. The standard mechanism is an efficient way to inhibit serine proteases, and it is thus used by many structurally disparate protein scaffolds to create potent inhibitors. However, most standard mechanism protease inhibitors tend to have relatively broad specificity within subclasses of serine proteases. For example, the bovine pancreatic trypsin inhibitor (BPTI) efficiently inhibits almost all trypsin-fold serine proteases with P1-Arg specificity, and it can also inhibit chymotrypsin (Phe P1 specificity) with a KI of 10 nM (22).
Standard mechanism inhibitors are classified strictly as inhibitors of serine proteases. There have been reports of inhibitors of other classes of proteases that have similar mechanisms to those of standard mechanism inhibitors, though. Initial studies on the streptomyces metalloprotease inhibitor (SMPI) suggest that it inhibits the metalloprotease thermolysin through a substrate-like binding mechanism (2). Similarly, staphostatin B, a cysteine protease inhibitor from Staphylococcus aureus, binds in a substrate-like manner in the active site of staphopain cysteine proteases. However, staphostatin B has a glycine P1 residue, which adopts a backbone conformation that seems to prevent nucleophilic attack of the scissile bond (3).

Figure 2. Standard mechanism of protein serine protease inhibitors bind in a substrate-like manner that completely spans the active site, and act as substrates with a very slow kcat. They interact with both the substrate binding sites (shallow indentation) and the catalytic residues (rectangle) of the serine protease.
Noncanonical competitive inhibitors
Several protease inhibitors bind in the active site of the protease, but they do not bind in a substrate-like manner, instead forming interactions with the catalytic residues in a noncatalytically competent manner, and thus, they are not considered standard mechanism inhibitors.
The cystatins, which are a superfamily of proteins that inhibit papain-like cysteine proteases, are a classic example of these inhibitors. The cystatins (Fig. 3) insert a wedge-like face of the inhibitor that consists of the protein N-terminus and two hairpin loops into the V-shaped active site of a cysteine protease. The N-terminal residues bind in the S3-S1 pockets in a substrate-like manner, but the peptide then turns away from the catalytic residues and out of the active site. The two hairpin loops bind to the prime side of the active site, which provides most of the binding energy for the interaction. Thus, both the prime and the nonprime sides of the active site are occupied, but no interactions are actually made with the catalytic machinery of the enzyme (23).
Tissue inhibitors of metalloproteases (TIMPs)
TIMPs inhibit matrix metalloproteases (MMPs) via a two-step mechanism in a manner somewhat similar to that of cystatins (Fig. 3). While the N-terminal residues of cystatins bind to the nonprime side of cysteine proteases, TIMPs N-termini bind in the P1-P3' pockets of the protease, coordinate the catalytic Zn2+ ion, and exclude a catalytic water molecule from the active site. Meanwhile a second loop of the TIMP binds in both the P3 and the P2 pockets, and it binds to the N-terminus of the MMP. Despite the similarities in mechanistic architecture between TIMPs and cystatins (hairpin loops and N-terminal residues in substrate binding pockets), TIMPs interfere with the catalytic machinery of MMPs by chelating the catalytic Zn2+ (5).
The ascaris pepsin inhibitor-3 is an aspin, which is a family of inhibitors of aspartic proteases that protect worms from host gastric enzymes. Like the cystatins, the aspins are competitive inhibitors that bind in the substrate-binding subsites, but they do not have an amide bond that is available for nucleophilic attack. They gain most of their inhibitory activity by inserting their 3 N-terminal residues in the S1'-S3' subsites of the protease (6) (Fig. 3).
Although cystatins and aspins do not interact directly with the catalytic residues of cysteine proteases, many protease inhibitors, such as cytotoxic T-lymphocyte antigen 2-α and the cathepsin propeptides, do interact with the catalytic machinery of the enzyme, but they do so in a proteolyticaly noncompetent manner. These inhibitors have long lengths of peptides that span the active site cleft in the reverse orientation (from C-terminus to N-terminus), thus preventing the catalytic cysteine residue from obtaining proper geometry for effective nucleophilic attack (24) (Fig. 3).

Figure 3. Competitive, active site inhibitors of proteases. These inhibitors bind in the active site, but not in a substrate-like manner. Peptide extensions bind in specificity subsites, and sometimes interact with the catalytic residues (rectangle), but not in a catalytically competent manner.
Competitive inhibition with exosite binding
Several protease inhibitors are competitive, and they bind in the protease active site, but also they have secondary binding sites outside the active site, which are critical to inhibition. Exosite binding provides two major benefits: 1) It increases the surface area of the interaction, which leads to a greater affinity, and 2) it can provide a greatly increased amount of specificity.
Ecotin is a dimeric serine protease inhibitor found in E. coli, which effectively inhibits many trypsin fold serine proteases, regardless of primary specificity, and is thought to protect E. coli from attack by host proteases. It inhibits serine proteases through a standard mechanism at a primary binding site, but it also has a secondary binding site that can contribute 5 kcal/mol of binding energy to the very tight enzyme-inhibitor complex (Fig. 4). Surprisingly, the individual binding energies of the two binding sites are not additive; the effect of the secondary binding site on affinity was found to be inversely proportional to the strength of binding at the primary site. The secondary binding site seems to provide compensatory effects that can overcome suboptimal binding at the primary binding site; if binding at the primary site is not optimal, the secondary binding interaction tends to be stronger. In this case, the secondary binding site actually makes the inhibitor less specific, or more capable of inhibiting a broad range of proteases, and it protects bacteria from several host proteases (25).
Many blood-meal parasites have developed specific inhibitors of clotting enzymes to prevent blood clotting of the host. These inhibitors often use mechanisms of inhibition described above, but they have domains that bind to protease exosites and provide a high degree of target specificity. Rhodniin, which is a thrombin inhibitor from the assassin bug Rhodnius prolixus, has two Kazal-type inhibitory domains and a common standard mechanism serine protease inhibitor domain. Although the N-terminal domain binds and inhibits via the standard mechanism, the second Kazal-type domain has evolved to bind to exosite I on thrombin. The binding affinities of the individual domains are roughly additive, and the resultant inhibitor has a KI of 0.2 pM and exquisite specificity for thrombin (26). Hirudin, from Hirudo medicinalis, and tick anticoagulant peptide (TAP) from Ornithodoros moubata, specifically inhibit thrombin and factor Xa (fXa), respectively (8, 27). They do so by similar mechanisms; they insert the N-terminal tail of the protein in the protease active site (analogous to aspin inhibition of pepsin), whereas the body of the inhibitor binds to specific exosites, either exosite I on thrombin (Fig. 4) or the autolysis loop on fXa. Because these secondary binding sites are specific to each clotting factor, the inhibitors show a high degree of specificity.
Inhibitor of apoptosis (IAP) proteins inhibit caspases, which are dimeric cysteine proteases responsible for programmed cell death, or apoptosis. IAPs are multidomain proteins that have multiple BIR domain repeats. One family member, the X-linked IAP (XIAP), has three BIR domains, and uses different BIR domains to inhibit different caspases through disparate mechanisms.
The XIAP-BIR2 domain and its N-terminal peptide extension are responsible for inhibition of the “executioner” caspases-3 and caspase-7. The N-terminal peptide extension binds in the active site in a reverse orientation, which is similar to the inhibition mechanism of cathepsin propeptides. Meanwhile, the BIR2 domain binds to an exosite on the caspase dimer (Fig. 4). The BIR2 domain needs both exosite binding capability and the N-terminal extension to inhibit efficiently its target caspases (28).
The XIAP-BIR3 domain is responsible for inhibition of the initiator caspase-9, but it functions via a completely different mechanism. The BIR3 domain is an allosteric inhibitor of caspase-9; it binds to the dimer interface and prevents dimerization and subsequent activation of the enzyme (9) (Fig. 4). Caspase-9 is at the apex of the apoptotic cascade that leads to the activation of executioner cascades. As such, BIR3 can provide an extra level of regulation by sequestering monomers in a catalytically inactive conformation and ensuring that no unwanted caspase-9 activity occurs.

Figure 4. Inhibitors that take advantage of exosite binding. These inhibitors are all competitive inhibitors that prevent substrate binding at the active site. BIR 3 prevents substrate binding because the active site is not formed in the caspase monomer. Exosite binding improves the potency and specificity of protease inhibitors.
Sometimes called suicide substrates, several protein inhibitors of proteases require proteolytic activity of the enzymes they inhibit, which leads to either covalent modification of the enzyme or releases charged groups that inhibit the catalytic machinery. In either case, this sort of activity-dependent inhibition is powerful and fundamentally different than the competitive mechanisms outlined above; the inhibitor acts as a substrate and then uses the enzymes’ catalytic machinery to trap and then inhibit the enzyme.
The potato metallocarboxypeptidase inhibitor (and metallo-carboxypeptidase inhibitors from leeches and ticks) inhibit carboxypeptidase B after a proteolytic processing event. These inhibitors bind their four C-terminal residues in the protease subsites S3-S1'. The C-terminal residue, Gly39, is processed, but it does not diffuse from the active site. In a type of product inhibition, Gly39 instead stays in the S1' pocket and chelates the catalytic Zn2+, which creates a protease-activated reversible inhibitor (29).
The inhibitor α-2-macroglobin and its relatives are responsible for clearing excess proteases from plasma. Less an inhibitor than a “protease sponge,” α2 M is a large protein, which is a tetramer of about 600 kD that has four bait loops on its surface. When a protease cleaves one of these reactive loops, it triggers a conformational change, and the protease becomes cross-linked to the inhibitor through surface lysines and arginines. The enzyme is still active; small-molecule substrates can still be hydrolyzed by proteases complexed with α2 M, but protein substrates are occluded from the active site and the complex is cleared quickly from the blood (30).
The serpins are a family of inhibitors that inhibit covalently and irreversibly primarily serine proteases (1) (the serpin Crm1 inhibits cysteine proteases). Serpins have a large reactive center loop (RCL) that is presented to a protease for proteolytic processing. During productive cleavage of the RCL, the N-terminal half of the RCL, which is still attached to the protease as an acyl-enzyme intermediate, is inserted into a P-sheet in the body of the inhibitor. The resulting free-energy change is enough to translocate the protease (which is still covalently attached to the RCL) to the distal side of the inhibitor, and the resulting steric collisions completely deform the protease active site, which thus leaves the protease tethered to the serpin and completely inactive (Fig. 5). The serpin inhibitory mechanism is completely irreversible. Because of the drastic nature and irreversibility of this mechanism, serpins function as protease scavengers, which protect cells and tissues from unwanted proteolytic activity.
These types of inhibitors, which take advantage of the catalytic activity of a protease to trap and inhibit the enzyme, are effective and powerful inhibitors. As discussed in the following section, many small-molecule irreversible and protease-activated inhibitors have been developed that rely on the same fundamental mechanism of using enzyme activity to trap and inactivate a protease.

Figure 5. Serpins inhibit serine proteases by binding a reactive center loop in the active site, forming a covalent complex with the enzyme, undergoing a large conformational change, and irreversibly distorting the active site of the protease.
Mechanisms of Small-Molecule Protease Inhibitors
To develop both tools for chemical biology and possible drugs, significant effort has gone into the discovery and development of small-molecule protease inhibitors. The two critical components in the design of a protease inhibitor, and indeed any enzyme inhibitor, are potency and specificity. It has been challenging to create potent and specific protease inhibitors for clinical use because of the high degree of similarity among families of proteases, but even relatively nonspecific protease inhibitors have been invaluable in teasing apart the roles proteases play in complex biologic processes.
An effective strategy for developing protease inhibitors has been to take peptide substrates that target the active site and to turn them into inhibitors by interfering with the catalytic machinery of the enzyme (Fig. 6). Although sometimes hampered by specificity problems, these types of inhibitors can be useful biologic probes, can help validate an enzyme as a drug target, and can act as lead compounds for additional drug development. Transition-state analogs—inhibitors that stably adopt a conformation that mimics the transition state of an enzyme-substrate intermediate—have traditionally been an effective way to develop inhibitors. In addition, several irreversible inhibitors have been developed that bind covalently to cysteine, serine, and threonine proteases, which are proteases that form a covalent acyl-enzyme complex during peptide bond hydrolysis.
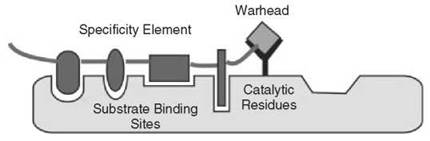
Figure 6. An effective strategy for developing synthetic protease inhibitors is to combine a peptide or peptidomimetic specificity element with a warhead that binds (either reversibly or irreversibly) to the catalytic machinery of a protease.
Transition-state inhibitors
Transition-state inhibitors stably mimic the transition state of the enzymatic reaction, and thereby interact with the substrate-binding and catalytic machinery of the enzyme in a low-energy conformation. Transition-state analogs are competitive, reversible inhibitors, although some have extremely low KI’s and very slow off-rates. All proteases activate a nucleophile to attack a carbonyl, which leads to the formation of a tetrahedral intermediate that then collapses to form the enzyme products—two peptides. Thus, synthetic small molecules that mimic the tetrahedral intermediate of the protease reaction are attractive transition-state analogs. A classic class of protease transition-state inhibitors uses a boronic acid scaffold (4, 10). Boronic acid adopts a stable tetrahedral conformation in the protease active site that is resistant to nucleophilic attack. Boronic acid inhibitors, which are derivatized with different specificity elements, have been developed against every class of protease and have been developed into therapeutic agents; the proteo-some inhibitor Bortezomib (Fig. 7) has been approved for the treatment of multiple myeloma (31).
Class-specific transition-state analogs have been developed to interfere specifically with the catalytic residues of each class of proteases. Aspartic protease inhibitors have long been designed around substrate polypeptides, with a replacement of the scissile amide bond with a noncleavable transition-state isostere. The first specific inhibitor for aspartic proteases, pepstatin A, was discovered from Actinomyces, as an inhibitor for pepsin. It also showed strong inhibitory activity against several other aspartic proteases. Pepstatin A is a peptide, but the scissile bond is replaced with a statine group [(3 S,4 S)-4-amino-3-hydroxyl-6-methyl heptanoic acid) (Fig. 7). Instead of a trigonal carbonyl, statines have a chiral hydroxyl group, which give it the ability to mimic the tetrahedral state of the substrate transition state (32). Other transition state isosteres, including homostatines, hydroxyethylenes, diols, and phosphinates have been developed for aspartic proteases, and functionalizing these classes of inhibitors with peptidomimetic groups to bind to the substrate binding regions, has resulted in the development of numerous drugs, most notably HIV protease drugs (33) (Fig. 7).
Another class of transition-state inhibitors is the peptide aldehyde inhibitors (Fig. 7). Aldehydes inhibit cysteine, serine, and threonine proteases via a covalent, reversible mechanism, and metalloproteases using an analogous but noncovalent mechanism. Aldehydes were discovered in screens for protease inhibitors from microorganisms and generally consist of a peptidyl moiety that binds in the non-prime specificity sites with a C-terminal aldehyde group. These inhibitors are substrate analogs that form a covalent hemiacetal linkage (or hemithio acetal linkage in cysteine proteases) between the aldehyde of the inhibitor and the active site nucleophile of the protease. The resulting tetrahedral adduct mimics the transition state of the normal enzymatic reaction. The tetrahedral intermediate can collapse and regenerate a free inhibitor; therefore, the enzyme-inhibitor complex is in equilibrium with a free enzyme and a free inhibitor. Many aldehyde inhibitors, such as leupeptin, are nonspecific and inhibit many proteases, but medicinal chemistry efforts taking advantage of unique protease specificity elements have led to several very specific aldehyde protease inhibitors (10).
Transition-state inhibitors, especially those with peptidyl or peptidomimetic extensions, are slow-binding inhibitors, and the protease-inhibitor binding mechanism includes one or more weakly bound intermediates before the formation of the tightly bound E•I complex. This slow-binding inhibition is a hallmark of inhibitors that bind in the active site in a substrate-like manner. In this way, transition-state analogs mimic the association mechanism of many of the naturally occurring protein inhibitors described above, particularly standard-mechanism inhibitors.

Figure 7. Various transition-state protease inhibitors. Bortezomib is an approved drug for the treatment of multiple myeloma. It is a boronic acid analog that inhibits the proteosome, a threonine protease. The boronic acid moiety can adopt a tetrahedral conformation in the active site. Pepstatin is a peptidyl aspartic acid inhibitor. The reactive statine group binds to the catalytic machinery, and the chiral hydroxyl group of the statine mimics the tetrahedral geometry of the transition state. Idinavir is an approved HIV 1 Protease inhibitor that binds to the active site via a hydroxyethylene transition state isostere. Aldehydes are also transition state analogs, which are susceptible to nucleophilic attack. In cysteine, serine and threonine proteases, this results in a covalent, reversible inhibition mechanism.
Irreversible inhibitors
Serine, cysteine, and theonine proteases, which perform a direct nucleophilic attack on the scissile bond (as opposed to the water-mediated nucleophilic attack performed by aspartic and metalloproteases) are excellent targets for covalent, irreversible inhibitors. Although pharmaceutical companies have been hesitant to pursue covalent inhibitors as drugs because of concerns over their possible cross-reactivity and potential for the development of an unwanted host immune response, it has been shown that a high degree of specificity can be built into these compounds. Furthermore, they are excellent biologic tools; when functionalized with fluorescent dyes or biotin, covalent inhibitors are effective imaging and pull-down reagents and they have been used to determine the presence and roles of active proteases in many biologic systems. Powers and coworkers (15) authoritatively reviewed these inhibitors, with detailed analysis of their mechanisms of action, structure-activity relationships, and their effectiveness in vivo.
Covalent irreversible inhibitors of cysteine, serine, and threonine proteases are capable of either alkylating or acylating their target enzymes (Fig. 8). Alkylating agents are very effective cysteine protease inhibitors, and they include chloromethyl ketones, fluoromethyl ketones, diazomethyl ketones, acyloxymethyl ketones, epoxides, and vinyl sulfones. The nucleophilic active site cysteine attacks an activated carbon and forms an irreversible carbon-sulfur bond. Because the active site serine of serine proteases is generally less nucleophilic than the corresponding catalytic cysteine, alkylating agents primarily target cysteine proteases. An exception is the chloromethyl ketones, which are capable of inhibiting serine proteases, although they have a slightly different mechanism of inhibition. After a nucleophilic attack of the carbonyl of the inhibitor, the inhibitor alkylates the catalytic histidine. The enzyme can then be deacylated, but the alkylation of the catalytic histidine results in a covalently bound inhibitor.
Inhibitors that acylate proteases present a carbonyl bond for nucleophilic attack, but because of either the poor electrophilicity of the acyl-enzyme intermediate or the inhibitor adopting a conformation unfavorable for deacylation, the enzyme reaction coordinate is trapped in the acyl-enzyme intermediate state. These inhibitors can inhibit both serine and cysteine proteases, and they can generally be sorted into two classes. Peptidyl-acylating agents have a peptide-like specificity element to target the inhibitor to a specific enzyme, but they have a modified scissile bond, such as an aza-group, a carbamate, or an acyl hydroxamate, that are resistant to deacylation. Several heterocyclic acylating agents also exist, such as isocoumarins, β-lactams, cephalosporins, and penems, which have their carbonyl groups enclosed in a ring structure. During nucleophilic attack, the ring is sprung open, and geometric or electrostatic effects can cause a stable acyl-enzyme complex.
Phosphonates (Fig. 8) and sulfonates represent a third class of covalent irreversible inhibitors. These inhibitors adopt a stable tetrahedral geometry and are covalently bound transition-state analogs. They often have a peptide-like specificity element, and the electrophilicity of the leaving groups can be modified to tune the reactivity of the inhibitor. These inhibitors are specific for serine proteases, because the serine protease active site has a well-defined oxyanion hole, which stabilizes the transition-state mimic.
A final group of covalent small-molecule inhibitors of proteases are mechanism-based inhibitors. These inhibitors are enzyme-activated irreversible inhibitors, and they involve a “two-hit” mechanism that completely inhibits the protease. Some isocoumarins and β-lactam derivatives have been shown to be mechanistic inhibitors of serine proteases. A classic example is the inhibition of elastase by several cephalosporin derivatives developed at Merck (Fig. 8). The catalytic serine attacks and opens the β-lactam ring of the cephalosporin, which through various isomerization steps, allows for a Michael addition to the active site histidine and the formation of a stable enzyme-inhibitor complex (34). These mechanism-based inhibitors require an initial acylation event to take place before the irreversible inhibitory event. In this way, these small molecules have an analogous mechanism of inhibition to the naturally occurring serpins and α-2-macroglobin, which also act as suicide substrates.

Figure 8. Irreversible inhibitors of proteases. Serine and cysteine proteases can be acylated by aza-peptides, which release an alcohol, but cannot be deacylated due to the relative unreactivity of the (thio) acyl-enzyme intermediate. Reactive carbons, such as the epoxide of E64, can alkylate the thiol of cysteine proteases. Phosphonate inhibitors form covalent bonds with the active site serine of serine proteases. Phosphonates are specific for serine proteases as a result of the rigid and well-defined oxyanion hole of the protease, which can stabilize the resulting negative charge. Mechanism-based inhibitors make two covalent bonds with their target protease. The cephalosporin above inhibits elastase [23]. After an initial acylation event that opens the β-lactam ring, there are a number of isomerization steps that eventually lead to a Michael addition to His57. Therefore, even if the serine is deacylated, the enzyme is completely inactive.
Reversible, competitive inhibitors
Concerns about toxicity, cross-reactivity, and immunogenicity have hampered the development of irreversible therapeutics that target proteases. Therefore, many noncovalent, reversible, competitive protease inhibitors have been developed. Most of these inhibitors interact with both the catalytic residues and the substrate binding sites, as binding to either individual element usually will not provide enough specificity or potency. In some cases, such as small-molecule peptidomimetic inhibitors of thrombin (35) (Fig. 9), binding to the substrate binding sites of the protease has provided sufficient potency, but this is likely because of the buried nature and unique specificity of the thrombin active site. In most cases, though, binding to the catalytic machinery provides potency, whereas the substrate binding sites provide opportunities for specificity. Metalloprotease inhibitors provide a representative example of reversible, competitive, small-molecule inhibitors. Metalloproteases have been the targets of multiple large-scale drug discovery efforts, and many inhibitors have been developed, with varying results. many inhibitors of the angiotensin-converting enzyme (ACE) have been brought to market to combat hypertension and myocardial infarction. Conversely, the first generation of matrix metalloprotease (MMP) inhibitors failed in clinical trials for the treatment of cancer, in part because the inhibitors had cross-reactivity with other metalloproteases. The common mechanistic feature between these inhibitors and, indeed, most small-molecule metalloprotease inhibitors is that they all have a functionality that chelates the catalytic zinc of the enzyme (36) (Fig. 9). Numerous chelating groups have been reported (37), such as hydroxamates, thiols, carboxylates, and boronic acids, but the challenge has been to develop molecules that both chelate the catalytic zinc and specifically bind to the substrate specificity regions of the enzyme. The second generation of MMP inhibitors have modified the reactivity of the metal-chelating group and have improved the specificity profiles of the substrate-binding moiety of the inhibitors (36). The difficulty in producing specific inhibitors might have been predicted by looking at naturally occurring macromolecular metalloprotease inhibitors. Four known human TIMPs exist, which are responsible for the inhibition of well over 20 MMPs. Clearly, MMP evolution has not been guided by the development of strict specificity elements. Instead, chelation of the catalytic zinc and substrate site binding defines naturally occurring metalloprotease inhibitors, and gaining specificity remains a difficult medicinal chemistry problem.

Figure 9.
As cataloged above, it can be difficult to develop specific protease inhibitors targeted to the active site of the enzyme, because of similarities in both the catalytic machinery and the binding sites among families of proteases. But as our understanding of protein dynamics grows, it is clear that allosteric regulation events control protease function, from zymogen activation, to dimerization, to substrate localization, to cofactor binding. Controlling these events offers a way to control protease activity more specifically. Inherent difficulties in this process exist, as these allosteric sites are often not well defined. Furthermore, it has proven difficult to interrupt protein-protein interactions with small molecules, as potential binding sites are usually shallow and solvent exposed. Despite these hurdles, recent strides have been made in the development of small-molecule allosteric inhibitors of proteases. To try to combat the rapid rate of resistance to HIV protease inhibitors, several groups have developed modified peptides that disrupt the HIV protease dimer and thus inactivate the enzyme (38). While searching for novel inhibitors of caspases, Hardy et al. (39) developed small-molecule inhibitors of caspase-3 and caspase-7. Crystal structures determined that the inhibitors bound near the dimer interface of the caspase-7, but they did not interfere with dimerization. They inhibited the enzyme by locking two loops forming the active site in a zymogen-like conformation that renders the enzyme catalytically inactive. This process is reminiscent of XIAP-BIR3 inhibition of caspase-9, which also locks the enzyme-active site in a catalytically inactive conformation by interfering with loop packing in the active site. Although relatively few small-molecule allosteric inhibitors of proteases have been discovered to this point, growing interest exists in allosteric regulation of proteases (as discussed in the following sections), and more allosteric inhibitors are sure to follow.
Inhibitors Developed via Protein Engineering
Protein engineering has allowed for the development of many new protease inhibitors with increased potency and specificity and for diverse mechanisms of action. Because of the relatively shallow active sites, homology, and broad specificity of many proteases, larger molecules are attractive inhibitors in that they can bury more surface area during binding and hopefully gain more potency and specificity.
One strategy has been to improve the specificity of naturally occurring protease inhibitors, either through rational design or via phage display. Mutations to residues that interact with the protease active site have drastic effects on inhibitor affinity, but specificity tends to be gained through evolution of secondary interactions. The standard mechanism serine protease inhibitors ecotin (40) and eglin c (41) have been refined at both of their primary and secondary interaction sites, which drastically improve their specificity for a single protease. Although not altering the primary mechanism of action of the inhibitors, the engineering of secondary binding sites gives these inhibitors a mechanism of inhibition similar to that of the anticlotting inhibitors such as hirudin, and greatly improves inhibitor specificity.
Another strategy has been to develop polypeptide-based inhibitors of proteases. Typically consisting of 10-20 amino acids, and often containing disulfide bonds to rigidify the inhibitors and decrease the entropic cost of binding, constrained peptides have been developed to inhibit aspartic, cysteine, serine, and threonine proteases. Although peptides are not ideal drug molecules because of their susceptibility to proteolysis, the relatively small size of constrained peptides allows for the creation of extremely diverse libraries. Furthermore, they are amenable to the incorporation of non-natural or D-amino acids, which thus greatly increases potential diversity. The mechanisms of action of these inhibitors have sometimes mimicked known biologic mechanisms, and sometimes they have been completely novel. Constrained peptide phage display libraries have yielded standard mechanism inhibitors of the serine proteases chymotrypsin (42) and urokinase-type plasminogen activator (uPA) (43) with moderate potency and specificity. Cyclic peptides have also been shown to inhibit competitively the aspartic protease renin, and they are thought to bind to the enzyme in a substrate-like manner (44).
Constrained peptides that mimic natural inhibitors are essentially a reduction of naturally occurring inhibitors to just their reactive elements. But several allosteric peptide inhibitors have been developed that have novel mechanisms of inhibition, which reveal information about enzyme function, and suggest new ways of regulating proteolysis. Constrained peptide libraries have yielded two extremely potent exosite inhibitors of the clotting enzyme factor Vila (fVIIa) (45, 46). The two inhibitors are bound to two different sites outside of the active site of the enzyme, and they had unique mechanisms of inhibition. One inhibitor, A-183, functioned by forcing a loop near the active site into an inactive conformation and by occluding substrate binding to the enzyme. The other inhibitor, E-76, was a noncompetitive inhibitor of FVIIa’s natural substrate, factor X, and it seems to work by locking the enzyme in a zymogen-like conformation. In another example of allosteric inhibition, an α-helical peptide was designed to disrupt—and thus prevent activation of—the dimerization of the protease from Kaposi’s sarcoma-associated herpes virus (KSHV) (47). The mechanisms of inhibition of these peptide inhibitors clearly overlap with those of both small-molecule and naturally occurring allosteric inhibitors; namely they lock their target enzymes in an inactive, closed, or zymogen state. The allure of allosteric inhibition is founded in the idea that it is possible to find multiple sites on an enzyme to regulate activity, and these molecules have done this by using established mechanisms to inhibit new proteases.
A third approach has been to mature specific protease inhibitors on other natural protein scaffolds, such as antibodies. Antibody inhibitors have been raised against metalloproteases (48), cysteine proteases (49), and serine proteases (50), and to this point, characterized inhibitors have either been monoclonal antibodies raised from hybridomas or from phage-display libraries. The mechanisms of inhibition are familiar; protease antibody inhibitors either interfere with multimerization (and thus activation) of a protease (51), bind to loops and protein-protein interaction sites to occlude substrate binding, or bind in the protease active site. The benefit of using antibodies as inhibitors is that they are exquisitely specific—antibodies have evolved to bind specifically to their antigen—and are useful biologic tools for imaging and in vivo experiments. And that they have been able to mimic the mechanism of inhibition of naturally occurring protease inhibitors suggests that specific antibody inhibitors can be developed for many proteases.
Conclusions and Future Directions
Relatively few design principles underlie the mechanisms of inhibition of an awe-inspiring range of protease inhibitors. Protease inhibitors tend to compete with substrate binding, either through direct competition or deformation of the protease active site. Although protein inhibitors can gain potency through the burial of a large surface area and specificity through contacts with specific exosites, small-molecule inhibitors primarily gain potency through interactions with the catalytic machinery of the enzyme and specificity through interactions with the substrate binding sites. The search for novel modes of enzyme control, such as allosteric regulation, is particularly exciting, with the hope that these regulatory sites will be more amenable to the design of specific inhibitors.
The design of new inhibitors based on these principles is critically important. A great deal remains to be learned about the role of proteases in biology, and effective, reliable chemical tools are needed to tease apart these processes. The failure of MMP inhibitors in clinical trials stemmed from toxicity problems (cross-reactivity that could be eliminated with more specific inhibitors), and just as critically, an incomplete understanding of which MMP was important to what stage of cancer growth and metastasis. A more thorough understanding of the role of individual proteases might have alleviated some of these issues. Because of their critical roles in biology, proteases are attractive drug targets. Although only a handful of anti-protease drug targets have approved inhibitors on the market thus far (DPPIV, HIV, ACE, and proteosome inhibitors), several drug discovery efforts that target proteases are underway, which aim to treat, for example, viral infections, parasitic infections, thrombosis, osteoporosis, neurodegenerative diseases, and cancer. As our understanding of the targets and the design principles of their inhibitors improve, more treatments are sure to follow (7, 12-14, 16-21).
References
1. Laskowski M Jr, Kato I. Protein inhibitors of proteinases. Annu. Rev. Biochem. 1980; 49:593-626.
2. Seeram SS, et al. Identification of reactive site of a proteinaceous metalloproteinase inhibitor from Streptomyces nigrescens TK-23. J. Biochem. 1997; 121:1088-1095.
3. Filipek R, Potempa J, Bochtler M. A comparison of staphostatin B with standard mechanism serine protease inhibitors. J. Biol. Chem. 2005; 280:14669-14674.
4. Rich DH. Inhibitors of cysteine proteinases. In: Proteinase Inhibitors. Barrett AJ, Salvesen G, eds. 1986. Elsevier, Amsterdam. pp. 153-178.
5. Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim. Biophys. Acta 2000; 1477:267-283.
6. Ng KK, et al. Structural basis for the inhibition of porcine pepsin by Ascaris pepsin inhibitor-3. Nat. Struct. Biol. 2000; 7:653-657.
7. McGrath ME, Gillmor SA, Fletterick RJ. Ecotin: lessons on survival in a protease-filled world. Protein Sci. 1995; 4:141-148.
8. Rydel TJ, et al. Refined structure of the hirudin-thrombin complex. J. Mol. Biol. 1991; 221:583-601.
9. Shiozaki EN, et al. Mechanism of XIAP-mediated inhibition of caspase-9. Mol. Cell 2003; 11:519-527.
10. Powers JC, Harper JW. Inhibitors of serine proteinases. In: Proteinase Inhibitors. Barrett AJ, Salvesen G, eds. 1986. Elsevier, Amsterdam. pp. 55-152.
11. Gettins PG. Serpin structure, mechanism, and function. Chem. Rev. 2002; 102:4751-4804.
12. Adams J, et al. Potent and selective inhibitors of the proteasome: dipeptidyl boronic acids. Bioorg. Med. Chem. Lett. 1998; 8:333-338.
13. Korting HC, et al. Effects of the human immunodeficiency virus (HIV) proteinase inhibitors saquinavir and indinavir on in vitro activities of secreted aspartyl proteinases of Candida albicans isolates from HIV-infected patients. Antimicrob. Agents Chemother. 1999; 43:2038-2042.
14. Umezawa H. Low-molecular-weight enzyme inhibitors of microbial origin. Annu. Rev. Microbiol. 1982; 36:75-99.
15. Powers JC, et al. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem. Rev. 2002; 102:4639-4750.
16. Oleksyszyn J, et al. Novel amidine-containing peptidyl phosphonates as irreversible inhibitors for blood coagulation and related serine proteases. J. Med. Chem. 1994; 37:226-231.
17. Doherty JB, et al. Cephalosporin antibiotics can be modified to inhibit human leukocyte elastase. Nature 1986; 322:192-194.
18. Gustafsson D, et al. The direct thrombin inhibitor melagatran and its oral prodrug H 376/95: intestinal absorption properties, biochemical and pharmacodynamic effects. Thromb. Res. 2001; 101:171 181.
19. Whittaker M, et al. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem. Rev. 1999; 99:2735-2776.
20. Cushman DW, et al. Design of potent competitive inhibitors of angiotensin-converting enzyme. Carboxyalkanoyl and mercaptoalkanoyl amino acids. Biochemistry 1977; 16:5484-5491.
21. Hooper NM, et al. Inhibition by converting enzyme inhibitors of pig kidney aminopeptidase P. Hypertension 1992; 19:281-285.
22. Castro MJ, Anderson S. Alanine point-mutations in the reactive region of bovine pancreatic trypsin inhibitor: effects on the kinetics and thermodynamics of binding to beta-trypsin and alpha-chymotrypsin. Biochemistry 1996; 35:11435-11446.
23. Bode W, Huber R. Structural basis of the endoproteinase-protein inhibitor interaction. Biochim. Biophys. Acta 2000; 1477:241-252.
24. Denizot F, et al. Novel structures CTLA-2 alpha and CTLA-2 beta expressed in mouse activated T cells and mast cells and homologous to cysteine proteinase proregions. Eur. J. Immunol. 1989; 19:631-635.
25. Eggers CT, et al. The role of ecotin dimerization in protease inhibition. J. Mol. Biol. 2001; 308:975-991.
26. Roussel A, et al. Complexation of two proteic insect inhibitors to the active site of chymotrypsin suggests decoupled roles for binding and selectivity. J. Biol. Chem. 2001; 276:38893-38898.
27. Wei A, et al. Unexpected binding mode of tick anticoagulant peptide complexed to bovine factor Xa. J. Mol. Biol. 1998; 283:147-154.
28. Scott FL, et al. XIAP inhibits caspase-3 and -7 using two binding sites: evolutionarily conserved mechanism of IAPs. EMBO J. 2005;24:645-655.
29. Arolas JL, et al. Secondary binding site of the potato carboxypeptidase inhibitor. Contribution to its structure, folding, and biological properties. Biochemistry 2004; 43:7973-7982.
30. Kolodziej SJ, et al. The three-dimensional structure of the human alpha 2-macroglobulin dimer reveals its structural organization in the tetrameric native and chymotrypsin alpha 2-macroglobulin complexes. J. Biol. Chem. 2002; 277:28031-28037.
31. Rajkumar SV, et al. Proteasome inhibition as a novel therapeutic target in human cancer. J. Clin. Oncol. 2005; 23:630-639.
32. Rich DH, Bernatowicz MS. Synthesis of analogues of the carboxyl protease inhibitor pepstatin. Effect of structure in subsite P3 on inhibition of pepsin. J. Med. Chem. 1982; 25:791-795.
33. Eder J, et al. Aspartic proteases in drug discovery. Curr. Pharm. Des. 2007; 13:271-285.
34. Navia MA, et al. Crystallographic study of a beta-lactam inhibitor complex with elastase at 1.84 A resolution. Nature 1987; 327:79-82.
35. Nilsson T, Sjoling-Ericksson A, Deinum J. The mechanism of binding of low-molecular-weight active site inhibitors to human alpha-thrombin. J. Enzyme Inhib. 1998; 13:11-29.
36. Fisher JF, Mobashery S. Recent advances in MMP inhibitor design. Cancer Metastasis Rev. 2006; 25:115-136.
37. Powers JC, Harper JW. Inhibitors of metalloproteases. In: Proteinase Inhibitors. Barrett AJ, Salvesen G, eds. 1986. Elsevier, Amsterdam. pp. 219-298.
38. Lee SG, Chmielewski J. Rapid synthesis and in situ screening of potent HIV-1 protease dimerization inhibitors. Chem. Biol. 2006; 13:421-426.
39. Hardy JA, et al. Discovery of an allosteric site in the caspases. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:12461-12466.
40. Stoop AA, Craik CS. Engineering of a macromolecular scaffold to develop specific protease inhibitors. Nat. Biotechnol. 2003; 21:1063-1068.
41. Komiyama T, et al. Optimization of protease-inhibitor interactions by randomizing adventitious contacts. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:8205-8210.
42. Dumez E, et al. Synthesis of macrocyclic, potential protease inhibitors using a generic scaffold. J. Org. Chem. 2002; 67:4882-4892.
43. Hansen M, et al. A urokinase-type plasminogen activator-inhibiting cyclic peptide with an unusual P2 residue and an extended protease binding surface demonstrates new modalities for enzyme inhibition. J. Biol. Chem. 2005; 280:38424-38437.
44. Nakaie CR, et al. Inhibition of renin by conformationally restricted analogues of angiotensinogen. Biochem. J. 1982; 205:43-47.
45. Roberge M, et al. A novel exosite on coagulation factor VIIa and its molecular interactions with a new class of peptide inhibitors. Biochemistry 2001; 40:9522-9531.
46. Dennis MS, et al. Peptide exosite inhibitors of factor VIIa as anticoagulants. Nature 2000; 404:465-470.
47. Shimba N, et al. Herpesvirus protease inhibition by dimer disruption. J. Virol. 2004; 78:6657-6665.
48. Matias-Roman S, et al. Membrane type 1-matrix metalloproteinase is involved in migration of human monocytes and is regulated through their interaction with fibronectin or endothelium. Blood 2005; 105:3956-3964.
49. Obermajer N, et al. Carboxypeptidase cathepsin X mediates beta2-integrin-dependent adhesion of differentiated U-937 cells. Exp. Cell Res. 2006; 312:2515-2527.
50. Petersen HH, et al. Localization of epitopes for monoclonal antibodies to urokinase-type plasminogen activator: relationship between epitope localization and effects of antibodies on molecular interactions of the enzyme. Eur. J. Biochem. 2001; 268:4430-4439.
51. Rezacova P, et al. Structural basis of HIV-1 and HIV-2 protease inhibition by a monoclonal antibody. Structure 2001; 9:887-895.
Further Reading
http://merops.sanger.ac.uk/ A comprehensive database of proteases and their inhibitors.
Barrett AJ, Salvesen G. Proteinase Inhibitors. Research Monographs in Cell and Tissue Physiology, Volume 12. 1986. Elsevier, New York.
Segel IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems. Wiley Classics Library ed. 1993. Wiley, New York.
Turk B. Targeting proteases: successes, failures and future prospects. Nat. Rev. Drug Discov. 2006; 5:785-799.
See Also
Approaches to Enzyme Inhibition
Aspartic Proteases and Aspartic Protease Inhibitors
Cysteine Proteases and Cysteine Protease Inhibitors
Metalloproteinases, Biophysics and Chemistry of
Serine Proteases and Serine Protease Inhibitors