CHEMICAL BIOLOGY
Unnatural Amino Acids to Investigate Biologic Processes
David B.F. Johnson*, Jeffrey K. Takimoto*, Jianfeng Xu* and Lei Wang, The Jack H. Skirball Center for Chemical Biology and Proteomics, The Salk Institute for Biological Studies, La Jolla, California
doi: 10.1002/9780470048672.wecb585
To circumvent the constraint imposed by the 20 canonical amino acids on the study of protein structure and function, various chemical and biosynthetic methods have been developed to incorporate unnatural amino acids into proteins. Unnatural amino acids now can be genetically encoded in living cells in a manner similar to that of common amino acids, which expands site-directed mutagenesis to diverse novel amino acids. The use of unnatural amino acids grants researchers a multitude of chemical and physical properties that cannot be found in the normal genetic repertoire, which significantly improves their ability to manipulate proteins and protein-involved biologic processes. Changes have been tailored into proteins to accurately dissect the contribution of hydrogen bonding, hydrophobic packing, cation-n interaction, and entropy to protein stability, as well as to precisely examine the structural and functional role of crucial residues. Unnatural amino acids also enable the introduction of new chemical reactivities, biophysical probes, mock posttranslational modifications, photoactive groups, and numerous other functionalities for the modification and regulation of protein activities. These studies not only reveal fundamental information of protein structure and function but also explore new means for generating novel protein properties and controlling biologic events.
Conventional site-directed mutagenesis of specific amino acids currently is the preferred method for investigating various structural and functional characteristics of proteins. A serious limitation to this methodology is the constraint of using the 20 canonical amino acids fixed by the universal genetic code. This constraint lies in the limited chemical and physical properties of these amino acids, which hinder the ability to make precise alterations. For instance, modification of an amino acid such as glutamine is limited because only asparagine has similar characteristics. For amino acids such as proline, no analogous amino acid exists in the genetic repertoire, which makes it difficult to investigate the role of this amino acid in specific processes without abolishing it completely. Breaking this limitation would enable in-depth investigation of the principles underlying protein structure and function as well as the engineering of novel protein properties and cellular functions. In the past decade, great progress has been made in incorporating unnatural amino acids into proteins to harness their extensive and powerful capabilities. Here, we will introduce unnatural amino acids with a brief overview of various methods for incorporating them into proteins and we will present examples that illustrate how unnatural amino acids have impacted a wide array of research that investigates biologic systems.
___________________________
* These authors contributed equally to this article.
Unnatural Amino Acids
Common amino acids consist of an amino group, a carboxyl group, a hydrogen atom, and a side chain all attached to the Ca in the L configuration. Analogs with altered side chains, or those that deviate from other features, are generally called unnatural amino acids (Fig. 1). The most widely used group of unnatural amino acids is the group in which the Ca side chain is changed. Variation of unnatural side chains is diverse and can range from structural analogs of canonical amino acids to those with specific chemical moieties, such as reactive functionalities and reporter groups for biophysical characterization. Modification of the amino group results in changes in the peptide backbone. For example, changing the amino group into a hydroxy or sulfhydryl group converts the endogenous amide bond between two residues into an ester and thioester link, respectively. Such changes make the resultant analog no longer an “amino” acid but an α-hydroxy acid and a thio acid. An analog with aminooxy replacing the amino group also has been incorporated into protein biosynthetically (1). The amino group also can be alkylated with different moieties to form amino acids that contain secondary amines (2). Another category of unnatural amino acids is α,α-disubstituted amino acids, whose α-hydrogen is replaced by an additional side chain. Moving the amino group away from the a carbon leads to extended β- or γ-amino acids, which can be compatible with protein biosynthetic machinery as well (3). Finally, D-amino acids, which are mirror images of the L counterparts, have been introduced selectively into functional proteins to study structural characteristics (4).

Figure 1. Different forms of unnatural amino acids.
Although this review is mainly focused on the use of unnatural amino acids in the investigation of biologic systems, a brief background of the methodology of incorporation is useful in understanding the power and application of this technology. A more comprehensive coverage of various methods can be found in Reference 5 and the references contained therein.
Chemical approaches
Global alterations to certain amino acids can be done in vitro through chemical modification of their exposed reactive side chains (6). The selectivity of chemical modification relies on the differences in chemical reactivity of amino acid functional groups. Judiciously selected chemicals will react with specific amino acids only, which allows chemical changes to be applied to that amino acid alone. Typical modification involves the thiol group of Cys, the ε-amino group of Lys, the carboxylate group of Asp and Glu, and the N-terminal amino group. The hydroxy group of Ser and Thr can be oxidized selectively when Ser and Thr are at the N-terminus of the protein (7). Side chains of Tyr and Trp can be modified selectively with transition metal catalysts (8, 9). Initial applications of this method focused on determining the functional roles of certain amino acid species for biologic activity, but they have expanded to other applications, including biophysical probe tagging, chemical cross-linking, and the conjugation of various synthetic functionalities. However, site-specific alterations are difficult using this approach because the chemical will react with all accessible target amino acids if more than one such amino acid exists in the protein. In addition, chemical modification must be done in vitro, and affects only those side chains that are solvent-accessible.
Another method to introduce unnatural amino acids into a polypeptide chain is through complete chemical synthesis (10). The predominantly used method, stepwise solid-phase peptide synthesis (SPPS), attaches the C-terminal amino acid to a solid support, and amino acids are added one at a time to the N-terminus. A clear advantage of chemical synthesis is that it enables the accurate introduction of unnatural amino acids at any site in a protein. The number of unnatural amino acids that can be introduced is limited only to the size of the chain, and chains of entirely unnatural amino acids can be produced using this method. Chemical synthesis is useful particularly for the incorporation of isotopic labels and unnatural amino acids that are toxic to cells or incompatible with the translational machinery. However, construction of a polypeptide chain using even the most advanced chemical synthesis techniques is daunting when confronted with the construction of an entire protein, as these methods currently are limited to approximately 100 amino acids (10).
Semisynthetic protein ligation methods, in which two or more protein fragments of recombinant or synthetic origin are chemically ligated to make the full-length protein (11), overcome the size limitation of SPPS. Among these methods, the native chemical ligation strategy couples peptide fragments to form a native peptide linkage, which leaves no chemical artifacts behind (12, 13). The desired unnatural amino acid is introduced in the synthetic fragment by using chemical synthesis and thus is incorporated into proteins after ligation. Once this unnatural protein is folded, biochemical characterization of kinetic parameters and function can be performed. Peptide ligation in living cells is also possible. A synthetic fragment can be injected into cells to react with an endogenously produced protein fragment (14). This method, like SPPS, has the power to introduce various unnatural structures that are synthetically accessible. However, it requires appropriate sites for cleavage and ligation, and it becomes cumbersome for internal sites in large proteins. Microinjection of either the in vitro ligation product for in vivo studies or the synthetic fragment for in vivo ligation can be a drawback to this method as well.
Biosynthetic approaches
Methods that use the endogenous cellular machinery to introduce unnatural amino acids into proteins are not limited by protein size and will facilitate the investigation of biologic processes in vivo. A general in vitro biosynthetic method allows for the site-specific incorporation of unnatural amino acids into proteins (15). In this method, a suppressor tRNA is chemically acylated with an unnatural amino acid, and the codon of interest in the target gene is mutated to the amber stop codon, TAG. When added to cell extracts that support transcription and translation, the suppressor tRNA recognizes and selectively incorporates the attached unnatural amino acid in response to the UAG in the transcribed mRNA. Using this method, a variety of unnatural amino acids have been incorporated into proteins, regardless of position or protein size, and have been applied to a large number of problems in protein chemistry (16). Besides the amber stop codon, rare codons and extended codons also have been used to specify the unnatural amino acid (17). An extension of this method involves the microinjection of the chemically acylated tRNA and UAG-containing mutant mRNA into Xenopus oocytes (18). The endogenous oocyte protein synthesis machinery supports translation and incorporation of the unnatural amino acid. This method enables the structure-function studies of integral membrane proteins, which are generally not amenable to in vitro expression systems (19). A purified in vitro translation system that consisted of only ribosomes, initiation factors, elongation factors, mRNA, and tRNAs preloaded with desired amino acids was used to incorporate simultaneously several unnatural amino acids into peptides in response to sense codons (20). By reassigning the meaning of codons, this system ultimately may allow the synthesis of peptides and proteins that contain multiple unnatural amino acids. The drawback to these methods lies in the chemical acylation of the suppressor tRNA, which is technically demanding and can exclude certain unnatural amino acid from attachment. In addition, acylated tRNA is consumed stoichiometrically and cannot be regenerated in cells or cell extracts, which leads to low expression of the target protein.
Multisite incorporation of unnatural amino acids by using cellular machinery has been achieved in auxotrophic bacterial strains (21) and in mammalian cells (22). This method relies on the idea that aminoacyl-tRNA synthetases, although with very high substrate specificity can mischarge unnatural amino acids that are close structural analogs of the cognate amino acids. An unnatural amino acid analogous to a canonical counterpart is introduced into a bacterial strain that is incapable of producing the natural amino acid or into mammalian cells that are deprived of the natural amino acid. The translational machinery then replaces the natural amino acid with its analog in all proteins. The incorporation efficiency of unnatural amino acids can be improved by increasing the expression level of the synthetase (23) and by introducing mutations that relax the substrate specificity of the aminoacylation domain (24) or attenuate the proofreading function of the editing domain of certain synthetases (25). However, this strategy is limited because it is restricted to global replacement of one amino acid with an analog and does not allow specific single alterations with a specific protein.
It would be ideal to genetically encode an unnatural amino acid in a manner similar to that of common amino acids, enabling site-directed mutagenesis in living cells with unnatural amino acids. A general method to expand the genetic code to include unnatural amino acids was developed. It involves the generation of a new tRNA-codon-synthetase set that is specific for the unnatural amino acid and does not crosstalk with other sets for common amino acids (26). The new synthetase is evolved to charge specifically an unnatural amino acid onto the new tRNA. This tRNA recognizes a codon that does not encode any common amino acids (e.g., a stop codon or an extended codon). When expressed in cells, the new tRNA-synthetase pair enables the unnatural amino acid to be site-specifically incorporated into proteins at the unique codon with high fidelity and efficiency. This method allows the use of unnatural amino acids in the investigation of biologic systems in an in vivo setting. It may be possible to generate stable cell lines or transgenic animals capable of inheriting such alterations for long-term studies. However, toxic unnatural amino acids and those incompatible with the protein biosynthesis machinery cannot be incorporated using this approach.
Application of Unnatural Amino Acids
Unnatural amino acids enable the structural, chemical, and physical properties of the building blocks of proteins to be customized according to needs. Such tailored changes have contributed to our understanding of the fundamental questions of protein chemistry on the molecular and atomic level, have been used to modify and enhance protein properties, and are being exploited to control protein activities to investigate various biologic processes and to create novel biologic functions.
Protein stability
There are many factors contributing to protein stability, including hydrogen bonding, hydrophobicity, packing, and conformational entropy, among others. It is difficult to access individual contributions by using conventional mutagenesis because changing one common amino acid to another often alters several properties at a time. For example, mutagenesis to disrupt hydrogen bonds, usually by deleting one member of a hydrogen-bonded pair, will leave an unpaired hydrogen donor or acceptor and/or alter local solvation and packing interactions, all of which may lead to protein destabilization.
To determine the effect of side-chain hydrogen bonding on protein folding, Tyr27 in staphylococcal nuclease (SNase) was replaced with several isosteric, fluorinated tyrosine analogs (unnatural amino acids 1 to 3) (Fig. 2) (27). These unnatural amino acids were designed to gradually increase the strength of the Tyr27-Glu10 hydrogen bond while minimizing the steric and electronic perturbations associated with deleting one hydrogen-bonding member. The stability constants Kapp of the corresponding mutants were found correlative with the pKa of the hydroxyl group in the tyrosine analogs. This result provides strong evidence that intramolecular side-chain hydrogen bonds preferentially stabilize the folded state of a protein relative to the unfolded state in water.
α-Hydroxy acids have been used to study the contribution of the backbone hydrogen bonds to protein stability (Fig. 3). The replacement of a common amino acid with an α-hydroxy acid that contains the same side chain effectively substitutes a good hydrogen-bond acceptor (the amide carbonyl group) with a considerably weaker one (the ester carbonyl group) in a conservative manner and disrupts a potential backbone hydrogen bond because the ester link cannot serve as a hydrogen-bond donor as does the NH. α-Hydroxy acids were incorporated at the N-terminus, the middle, and the C-terminus of the a-helix 39-50 of T4 lysozyme (28). At the N-terminus and the C-terminus, where only one hydrogen-bonding interaction is perturbed, the ester substitution destabilizes the protein by 0.9 kcal mol-1 and 0.7 kcal mol-1, respectively. In the middle of the helix, where such substitution perturbs two hydrogen bonds, the protein is destabilized by 1.7 kcal mol-1. In another study, Leu 14 in an antiparallel P sheet of SNase was replaced with leucic acid (29). This amide-to-ester change decreases the stability by 1.5—2.5 kcal mol-1. Altogether, these results convincingly show that both side-chain hydrogen bonds and main-chain hydrogen bonds significantly contribute to protein stability.
To examine the importance of the packing interaction in the core of a protein, Leu133 in T4 lysozyme was replaced with a series of analogs with extended or shortened alkyl side chains (unnatural amino acids 4 to 7) (30). Leu133 lies along the edge of the largest cavity in the interior of T4 lysozyme, which makes it possible to change the bulk of the side chain with minimal concomitant strain. Incorporation of (S,S)-2-amino-4-methylhexanoic acid (unnatural amino acid 4) and (S)-2-amino-4-cyclopentylpropanoic acid (unnatural amino acid 5) stabilizes T4 lysozyme by 0.6 kcal mol-1 and 1.24 kcal mol-1, respectively, which indicates that the increased bulk of buried hydrophobic residues can enhance protein stability. During protein folding, the cyclic amino acid 5 will lose less conformational entropy than does 4. That the 5-containing mutant is more stable than the 4-containing mutant suggests that side-chain entropy also affects protein stability. As expected, when the side chain of Leu133 is shortened systematically, as in unnatural amino acids 6, 7, and alanine, the protein becomes increasingly less stable.
Another method for increasing hydrophobicity while minimizing structural perturbation is to replace hydrocarbons with fluorocarbons. Using solid-phase synthesis, L-5,5,5,5',5',5'-hexafluoroleucine 8 was substituted for seven core leucine residues in a 30-residue peptide that can form homodimeric coiled coils. Hydrophobic side chains of the core residues pack against each other in the coiled coil. Fluorination of these side chains increased the hydrophobicity and raised the melting temperature of the homodimer from 34°C to 82° C (31). In addition, fluorocarbons are insoluble in hydrocarbons at room temperature and, thus, form a fluorous phase by interacting with other fluorocarbons. When a disulfide-bound heterodimer of the hexafluoroleucine core peptide and a leucine core peptide was allowed to undergo disulfide exchange, the peptides self-sorted into homodimers (31). This fluorous effect could lead to a novel protein-protein recognition. In another report, six leucine residues in the hydrophobic core of an antiparallel 4-α-helix bundle were replaced by 8 (32). The free energy of the unfolding of the mutant peptide increases by 0.3 kcal mol-1 per residue when the two central leucines are substituted and by an additional 0.12 kcal mol-1 per residue when the outer leucines are replaced, which confirms that hydrophobic packing stabilizes proteins.

Figure 2. Structures of unnatural amino acids discussed in the text.
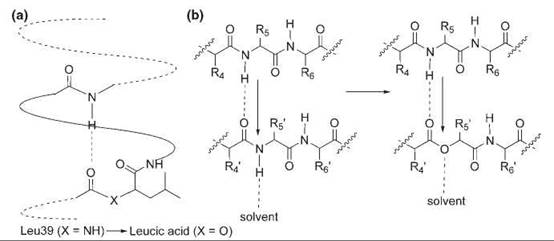
Figure 3. Backbone mutations generated by α-hydroxy acids. (a) N-terminal mutation of Leu39 to leucic acid in an α-helix of the T4 lysozyme. (b) Substitution of Leu14 with leucic acid in a β sheet of SNase.
Protein structure and function
Structural and Functional Role of Specific Residues
Unnatural amino acids can be designed to elucidate the functional role of a residue that is misinterpreted by or remains ambiguous to conventional mutagenesis and other methods. For example, Glu43 is important for the catalytic activity of SNase because its replacement by Asp and Gln significantly decreases the catalytic efficiency. Previous structural and mutagenesis studies suggested that Glu43 functions as a general base to activate a water molecule for hydrolyzing the phosphodiester bond of DNA. However, substitution of Glu43 with either homoglutamate (unnatural amino acid 9) or (S)-4-nitro-2-aminobutyric acid (unnatural amino acid 10) yielded mutant enzymes with kinetic constants similar to those of wide-type SNase (33). Because these two unnatural amino acids are isoelectronic and isosteric to glutamate but a much poorer base, such substitution would decrease SNase activity if Glu43 were a general base during catalysis. In addition, the X-ray crystal structure of the homoglutamate mutant showed that the carboxylate side chain of this residue occupies a position and orientation similar to that of Glu43 in the wild-type enzyme. Therefore, Glu43 may play a structural role instead and serve as a bidendate hydrogen-bond acceptor to fix the conformation of the neighboring loop.
Proline is unique among the natural amino acids in that its α-nitrogen is part of a pyrrolidine ring. The proline residue disrupts main-chain hydrogen bonding; it cannot serve as a hydrogen-bond donor because of the lack of a backbone NH moiety. Also, proline forms cis-peptide bonds at a frequency (5%) much higher than any other natural amino acids (<0.1%). In ion channels, Pro often is conserved at crucial sites, such as Pro221 in the nicotinic acetylcholine receptor (nAChR) and Pro256 in the 5-hydroxytryptamine-3A receptor (5-HT3AR). To probe which feature of Pro is functionally significant, a-hydroxyl acids (analogs of Gly, Val, and Leu) were incorporated at these sites, which all produced mutant receptors with properties similar to the wild-type receptor (34, 35). In contrast, incorporation of canonical amino acids Gly, Ala, or Leu yielded nonfunctional receptors. Because a-hydroxyl acids similarly lack the NH moiety for backbone hydrogen bonding and the nature of side chains does not affect receptor activity, these results suggest that the functional importance of the conserved Pro in both receptors is to remove backbone hydrogen bonding.
Another conserved proline residue of the 5-HT3AR, Pro308, has been shown to be indispensable for channel gating using conventional mutagenesis. However, substitution of this Pro with α-hydroxy acids produced nonfunctional receptors, which suggests that the lack of backbone hydrogen bonding is not the key to the proper function of this Pro. Interestingly, proline analogs that strongly favor the trans conformer (unnatural amino acids 11 and 12) produced no gating response, but those that favor the cis conformer (unnatural amino acids 13 and 14) yielded highly sensitive channels. Moreover, a linear energy correlation was observed between the cis-trans energy gap of the proline analogs and the receptor activation (36). This study strongly suggests that the critical role of Pro308 is to provide the switch that interconverts the open and closed states of the channel through cis-trans isomerization.
Cation-π Interaction
Cation-n interaction is a noncovalent electrostatic interaction between a cation and the electrons in n orbitals, which plays an important role in protein structure, binding, and catalytic function. The energetic contribution of this interaction to proteins cannot be measured accurately with conventional mutagenesis because no positively charged natural isosteres exist for common amino acids. To engineer a cation-π interaction in SNase, Val74, which occupies a hydrophobic pocket composed of one tyrosine side chain and two phenylalanine side chains, was replaced with the positively charged S-methylmethionine (unnatural amino acid 15). Another mutant was made by replacing Val74 with homoleucine (unnatural amino acid 16), which is isosteric to S-methylmethionine. Comparison of the thermodynamic stability of these two mutant proteins showed that the magnitude of cation-n interaction is about 2.6 kcal mol-1 (37).
A number of aromatic amino acids have been identified near the agonist-binding site of the nAChR, which suggests that cation-π interactions may be involved in binding the quaternary ammonium group of the agonist acetylcholine. A series of progressively fluorinated tryptophan derivatives (unnatural amino acids 17 to 20) were incorporated at aTrp149. Because fluorine is an electron-withdrawing group, substitution of H with F in the aromatic ring weakens the cation-π interaction. Ab initio quantum mechanics was used to predict the cation-π-binding abilities of the fluorinated tryptophans, and the calculated binding energy has a linear relationship with receptor activation by the agonist (38). Such correlations were not observed for other aromatic residues, which suggests that the cation-π interaction indeed exists for agonist binding and pinpoints it to αTrp149. This interaction was shown later as a general binding pattern between the Cys-loop superfamily of neurotransmitter receptors, such as the 5-HT3A receptors and the γ-aminobutyric acid receptors, and their cationic ligands or substrates (39).
Biophysical Probes
The site-specific introduction of biophysical probes into proteins has proven extremely powerful in revealing subtle changes of proteins with high spatial resolution. The carbon-deuterium (C-D) bond absorbs at ~2100 cm-1, which is within the transparent IR window (~1800-2700 cm-1) of proteins and, therefore, makes it easily observable by IR spectroscopy. The inherently fast timescale of IR spectroscopy also provides high temporal resolution. Therefore, unnatural amino acids with C-D bonds are excellent probes of protein folding and dynamics. Absorptions at different frequencies indicate the existence of multiple intermediates, and an increased line width of the absorption shows increased flexibility of the local environment. Amino acids containing C-D bonds were incorporated at different positions throughout cytochrome c (cyt c) by using semisynthetic approaches (40). By characterizing the absorption frequencies and line widths of the C-D bonds of these residues, it was found that no significant difference exists in the flexibilities of the oxidized and reduced states of cyt c. The data also show that parts of the protein exist in dynamic equilibrium with locally unfolded states and that cyt c is less stable than previous studies suggest.
Another infrared probe, p-cyano-L-phenylalanine (pCNPhe, 27), has been genetically encoded in E. coli and used to examine different ligand-bound states of the heme group in myoglobin (41). The stretching vibration of the nitrile group of p CNPhe has strong absorption and a frequency (vCN) at ~2200 cm-1, which falls in the transparent window of protein IR spectra. A substitution of pCNPhe was made for His64, which is at the distal face and close to the iron center of the heme group in myoglobin. In the ferric myoglobin, when the Fe(III) ligand was changed from water to cyanide, vCN shifted from 2248 cm-1 to 2236 cm-1, which indicates a less polar active site. In the ferrous myoglobin, a vCN absorption at 2239 cm-1 was observed for the linear Fe(II)CO complex, and the bent Fe(II)NO and Fe(II)O2 complexes showed a vCN absorption at 2230 cm-1. These results demonstrate that the nitrile group is a sensitive probe for ligand binding and for local electronic environment.
Small fluorescent probes sensitive to various environmental changes have the great potential for monitoring many biologic events as a complementary reporter for the widely used fluorescent proteins. For example, L-(7-hydroxycoumarin-4-yl)ethyl-lycine (CmrGly, 28) has been incorporated into holomyoglobin to study its local unfolding (42). CmrGly was incorporated at position Ser4 in helix A and at position His37 in helix C, respectively. The coumarin fluorescence intensity increases with solvent polarity. When the Ser4CmrGly mutant was unfolded with 2M urea, its fluorescence increased 30%, which indicates that helix A is disordered. In contrast, the fluorescence intensity of the His37CmrGly mutant did not change significantly until the urea concentration was raised to 3 M. These results suggest helix C and helix A unfold at different times and concentrations of the denaturing agent.
Modification and regulation of protein activity
Green fluorescent protein (GFP), whose chromophore is auto- catalytically formed by the tripeptide Ser65-Tyr66-Gly67, has become one of the most important in vivo markers for biologic studies. An aromatic amino acid at position 66 is necessary for fluorescence generation. To determine how the spectral properties of GFP could be altered by this residue, tyrosine analogs bearing different substituents at the para position of the phenyl ring (unnatural amino acids 21 to 24) were used to replace Tyr66 (43). The absorbance and fluorescence emission maxima of mutant GFPs are all blue-shifted, spanning the range from 375 to 435 nm and 428 to 498 nm, respectively. The wavelengths of the maxima increase in the order of bromo, iodo, methoxy, hydroxyl, amino, and deprotonated hydroxyl group. This shifting trend is consistent with the electron-donating ability of the substituents. In another experiment, Trp66 of the enhanced cyan fluorescent protein was replaced with L-4-aminotryptophan (unnatural amino acid 25) (44). The electron-donating amino group significantly red-shifts the fluorescence emission by 69 nm, which changes the color from cyan to gold.
Comparison of the p-methoxy-Phe (unnatural amino acid 22) mutant GFP with wild-type GFP also provides direct evidence for the peak assignment of GFP. Wild-type GFP has two absorbance maxima at 397 nm and 475 nm, which are believed to correspond to a neutral chromophore (phenol of Tyr66) and an anionic chromophore (phenolate anion of Tyr66), respectively. Excitation at either absorbance peak leads to a single fluorescence emission centered at 506 nm, which corresponds to the anionic chromophore in the excited state (45). Picosecond spectroscopy revealed that the excited neutral chromophore should emit at 460 nm (46). The absence of 460 nm emission in wild-type GFP suggests that an excited state proton transfer process is involved. Substitution of the hydroxyl group of Tyr with a methoxy group removes the possibility of deprotonation and proton transfer. Indeed, when Tyr66 is replaced with p-methoxy-Phe, only one absorbance maximum at 394 nm is observed, which is close to the absorbance maximum of the neutral chromophore of wild-type GFP. Moreover, only one emission maximum at 460 nm is detected for this mutant, which corroborates the ultrafast spectroscopic results (43).
The specificity of nucleic acid-binding proteins relies greatly on the hydrogen bonding between protein polar atoms and nucleic acid bases. Unnatural amino acids that can change isosterically the hydrogen-bonding pattern have been exploited to alter the substrate specificity. The λ-repressor recognizes the C:G pair at position 6 in the operator site OL1, and Lys4 of the λ-repressor is crucial for this recognition. The ε-NH2 group of Lys4 forms hydrogen bonds with the carbonyl group of Asn55 and the 6-oxo group of the guanine, functioning as as two hydrogen bond donors. Substitution of Lys4 with isosteric S-(2-hydroxyethyl)-cysteine changes the ε-NH2 to the -OH group, which now should accept hydrogen bonding from the amino group of adenine while preserving hydrogen bonding with Asn55 as a donor (Fig. 4). In fact, after the unnatural amino acid was introduced into the λ-repressor through site-directed mutagenesis and chemical modification, the binding specificity was switched from the C:G to T:A base pair (47).
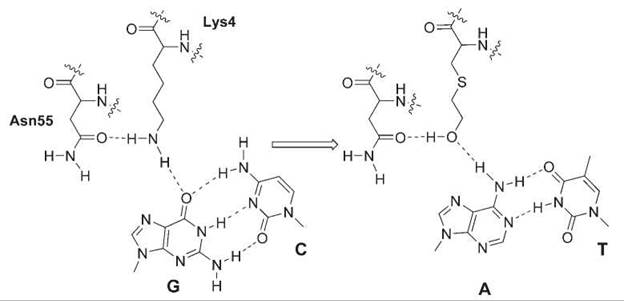
Figure 4. Substitution of Lys4 with 2-hydroxylethyl-cysteine in the λ-repressor changes the hydrogen-bonding pattern and DNA substrate specificity from C:G to T:A.
The chirality of D-amino acids has been harnessed for pharmaceutical purposes. D-peptide ligands should be resistant to proteolytic degradation and thus are more desirable as drugs. However, large libraries of D conformers cannot be encoded genetically and expressed for selection. A method termed mirror-image display solved this problem in an intriguing way (48). An L-peptide library is encoded genetically and displayed on the phage surface, and peptides of this library are selected by the target protein that is synthesized using all D-amino acids. The identified L-peptide then is resynthesized using D-amino acids, which should interact with the target protein of the natural handedness for reasons of symmetry. This approach has been used successfully to identify D-peptides that bind the Src homology 3 domain of c-Src and the HIV-1 gp41 protein (48, 49).
Unnatural amino acids that mimic posttranslational modifications can be used to control protein functions. For example, protein phosphorylation regulates many signal transduction pathways and is a reversible process catalyzed by various phosphatases and kinases. The dynamic change of the phosphorylation status of a protein makes it difficult to study the effect of this modification in detail. The generation of metabolically stable phosphoproteins would be useful to dissect the function and to direct signal transduction. Unnatural amino acid p-carboxymethyl-L-phenylalanine (pCMF, 26) is a nonhydrolyzable analog of phosphotyrosine and was found capable of mimicking the phosphorylated state of Tyr. This capability was demonstrated in a model phosphoprotein, the human signal transducer and activator of transcription-1 (STAT1). STAT1 has only a weak affinity for DNA, but during phosphorylation of Tyr701, STAT1 forms a homodimer and strongly binds a DNA duplex that contains M67 sites. The mutant STAT1 with Tyr701 substituted with pCMF also bound the M67-containing DNA duplex tightly, which suggests that p CMF could replace phosphotyrosine in the generation of constitutively active phosphoproteins (50).
The development of photoactive amino acids provides researchers with an extremely useful tool not only to probe biologic function but also to control spatially and temporally a variety of biologic processes. One strategy is to attach a suitable photoremovable protecting group to the amino acid, which renders the amino acid inactive. Photolysis releases the caging group and converts the amino acid to an active form, which generates abrupt or localized changes to the target protein. The 2-nitrobenzyl derivative is the most prevalent form for caged compounds. For example, the conserved Ser1082 at the upstream splice junction of the self-splicing DNA polymerase of Thermococcus litoralis was substituted with o-(2-nitrobenzyl)serine (Fig. 5a). The full-length precursor protein underwent protein splicing only when the unnatural residue was reverted back to wild-type Ser during photolysis (51). In other examples, o-nitrobenzyltyrosine (Fig. 5b) was used to replace Tyr93 or Tyr198 in the α subunit of the nAChR. These two Tyr residues are highly conserved for agonist binding. Millisecond flashes of light at 300-350 nm decaged the protected tyrosines and produced abrupt increments of currents that were conducted by the ion channel (52). Also, o-nitrobenzyltyrosine has been incorporated at the essential Tyr503 site of β-galactosidase to activate its enzymatic activity by using light both in vitro and in E. coli (53). Mutation of the active-site cysteine residue in the proapoptotic protease caspase 3 to o-nitrobenzylcysteine led to a catalytically inactive enzyme, whose activity could be restored by photocleavage (54). In addition to caging the active side chains, the 2-nitrobenzyl group has been harnessed also to cleave the protein backbone photochemically. 2-Nitrophenyl glycine (Fig. 5c) was introduced into sites of the signature disulfide loop of the nAChR. Irradiation at 360 nm resulted in site-specific backbone lesion and an almost complete loss of nAChR activity (55).

Figure 5. Photolysis of 2-nitrobenzyl caged serine (a) and tyrosine (b) restores the wild-type residues. Photolysis of 2-nitrophenyl glycine (c) cleaves the protein backbone.
Photolysis of a caged amino acid residue is an irreversible process. Reversible modulation can be achieved with the photochromic azobenzene compounds. Azobenzene undergoes a reversible cis-trans isomerization: The more stable trans isomer can be converted to the cis isomer upon illumination at 320-340 nm, and the cis-form can revert to trans-form either thermally or by irradiation at >420 nm. The resultant change in geometry and/or dipole of the compound can be used for regulating protein activity. For example, a known K+ channel blocker, tetra-ethyl ammonium, was linked via an azobenzene group to a cysteine that was introduced at specific sites of a K+ ion channel (Fig. 6a). When the azobenzene group isomerizes between the extended trans -form and the shorter cis-form in response to specific wavelengths of light, the structural change moves the blocker into or out of channel-blocking position and, thus, opens and closes the ion channel, respectively (58). Such photomodulation can be used to control neuronal activity noninvasively. The azobenzene group has been genetically encoded in the form of phenylalanine-4'-azobenzene (AzoPhe). AzoPhe was incorporated at the Ile71 site of the E. coli catabolite activator protein, a transcriptional activator. Its binding affinity for the promoter sequence decreased fourfold after irradiation at 334 nm (Fig. 6b), which converts the predominant trans AzoPhe to the cis-form. The isomerized cis AzoPhe then was switched back to the trans-state by irradiation at >420 nm, after which the affinity of the protein for the promoter was completely recovered (56).

Figure 6. (a) The geometrical change resultant from the cis-trans isomerization of azobenzene moves an ion channel blocker in and out of the ion channel to close and open the ion channel, respectively. Such activity was used to modulate the spontaneously firing hippocampal neurons. The firing frequency is significanly decreased when the azobenzene is in the cis-form after irradiation at 390 nm. Normal firing behavior is restored during irradiation at 500 nm (reprinted from (57), Copyright 2005, with permission from Elsevier). (b) Structure of phenylalanine-4'-azobenzene (AzoPhe) in trans-form and gel mobility shift assay to determine the binding affinity of the catabolite activator protein (CAP) to the lactose promoter DNA fragment (reprinted with permission from (56), Copyright 2006, American Chemical Society). Lane 1, DNA only. Lane 2, DNA+wild-type CAP. Lane 3, DNA+CAP with AzoPhe incorporated at residue 71 (after irradiation at 334 nm). Lane 4, DNA+CAP with AzoPhe incorporated at residue 71 (before irradiation at 334 nm). Substitution of Ile71 with trans AzoPhe in CAP results in a fourfold decrease of the binding constant Kb of the CAP for its promoter sequence. Photoirradiation at 334 nm partially converts the trans AzoPhe to the cis-form and decreases the Kb by another fourfold. The latter affinity loss can be completely recovered after irradiation at > 420 nm, which switches the cis-form back to the predominant trans-state.
The examples summarized here are only representative and by no means comprehensive. Many unnatural amino acids now can be incorporated, but simply have yet to be used in the investigation of biologic function. Unnatural amino acids that contain photocross-linkers, biophysical probes, chemical moieties with unique reactivities, and posttranslational modifications, among many others, have much promise in their capabilities. The use of these amino acids will expand the capabilities of probing protein structure and function as well as protein-involved biologic processes. The methodology of incorporation is advancing as well. It may be possible to genetically encode unnatural amino acids in many other cell types and organisms. The incorporation of multiple unnatural amino acids simultaneously by using extended codons may enable more complex investigations to be performed.
Additional work using unnatural amino acids can lead to the design and synthesis of novel and diverse biologic functions. By incorporating specific chemical moieties and physical characteristics into proteins, new protein properties may be discovered and used. Such exploration can be attempted either rationally or combinatorially. Diversities of protein libraries would be increased greatly by the addition of only a few unnatural amino acids, which may enhance the probability of discovering proteins that contain novel properties and functions. It is easy to see how unnatural amino acids can be extended into the pharmaceutical industry to create more efficient therapeutics. Finally, the creation of a sustainable organism that is capable of using unnatural amino acids will enable the investigation of the evolution of the genetic code on this planet.
Acknowledgments
We thank A.R. Parrish for help editing this manuscript. J. Xu is supported in part by the Pioneer Fellowship of the Salk Institute. L. Wang is a Searle Scholar and a Beckman Young Investigator.
References
1. Eisenhauer BM, Hecht SM. Site-specific incorporation of (amino-oxy)acetic acid into proteins. Biochemistry 2002; 41:11472—11478.
2. Ellman JA, Mendel D, Schultz PG. Site-specific incorporation of novel backbone structures into proteins. Science 1992; 255:197-200.
3. Hartman MC, Josephson K, Szostak JW. Enzymatic aminoacylation of trna with unnatural amino acids. Proc. Natl. Acad. Sci. U. S. A. 2006; 103:4356-4361.
4. Valiyaveetil FI, Sekedat M, Mackinnon R, Muir TW. Glycine as a d-amino acid surrogate in the k(+)-selectivity filter. Proc. Natl. Acad. Sci. U. S. A. 2004; 101:17045-17049.
5. Wang L, Schultz PG. Expanding the genetic code. Angew. Chem. Int. Ed. Engl. 2004; 44:34-66.
6. Means GE, Feeney RE. Chemical modifications of proteins: history and applications. Bioconjug. Chem. 1990; 1:2-12.
7. Geoghegan KF, Stroh JG. Site-directed conjugation of nonpeptide groups to peptides and proteins via periodate oxidation of a 2-aminoalcohol: Application to modification at n-terminal serine. Bioconjug. Chem. 1992; 3:138-146.
8. Antos JM, Francis MB. Selective tryptophan modification with rhodium carbenoids in aqueous solution. J. Am. Chem. Soc. 2004; 126:10256-10257.
9. Tilley SD, Francis MB. Tyrosine-selective protein alkylation using pi-allylpalladium complexes. J. Am. Chem. Soc. 2006; 128:1080-1081.
10. Kent SB. Chemical synthesis of peptides and proteins. Annu. Rev. Biochem. 1988; 57:957-989.
11. Wallace CJ. Peptide ligation and semisynthesis. Curr. Opin. Biotechnol. 1995; 6:403-410.
12. Dawson PE, Muir TW, Clarklewis I, Kent SBH. Synthesis of proteins by native chemical ligation. Science 1994; 266:776-779.
13. Muir TW, Sondhi D, Cole PA. Expressed protein ligation: a general method for protein engineering. Proc. Natl. Acad. Sci. U. S. A. 1998; 95:6705-6710.
14. Giriat I, Muir TW. Protein semi-synthesis in living cells. J. Am. Chem. Soc. 2003; 125:7180-7181.
15. Noren CJ, Anthony-Cahill SJ, Griffith MC, Schultz PG. A general method for site-specific incorporation of unnatural amino acids into proteins. Science 1989; 244:182-188.
16. Cornish VW, Mendel D, Schultz PG. Probing protein structure and function with an expanded genetic code. Angew. Chem. Int. Ed. Engl. 1995; 34:621-633.
17. Hohsaka T, Ashizuka Y, Taira H, Murakami H, Sisido M. Incorporation of nonnatural amino acids into proteins by using various four-base codons in an escherichia coli in vitro translation system. Biochemistry 2001; 40:11060-11064.
18. Nowak MW, Kearney PC, Sampson JR, Saks ME, Labarca CG, Silverman SK, Zhong WG, Thorson J, Abelson JN, Davidson N, Schultz PG, Dougherty DA, Lester HA. Nicotinic receptor binding site probed with unnatural amino acid incorporation in intact cells. Science 1995; 268:439-442.
19. Dougherty DA. Unnatural amino acids as probes of protein structure and function. Curr. Opin. Chem. Biol. 2000; 4:645-652.
20. Forster AC, Tan Z, Nalam MN, Lin H, Qu H, Cornish VW, Black- low SC. Programming peptidomimetic syntheses by translating genetic codes designed de novo. Proc. Natl. Acad. Sci. U. S. A. 2003; 100:6353-6357.
21. Budisa N. Prolegomena to future experimental efforts on genetic code engineering by expanding its amino acid repertoire. Angew. Chem. Int. Ed. Engl. 2004; 43:6426-6463.
22. Suchanek M, Radzikowska A, Thiele C. Photo-leucine and photomethionine allow identification of protein-protein interactions in living cells. Nat. Methods 2005; 2:261-267.
23. Tang Y, Tirrell DA. Biosynthesis of a highly stable coiled-coil protein containing hexafluoroleucine in an engineered bacterial host. J. Am. Chem. Soc. 2001; 123:11089-11090.
24. Ibba M, Kast P, Hennecke H. Substrate specificity is determined by amino acid binding pocket size in escherichia coli phenylalanyl-trna synthetase. Biochemistry 1994; 33:7107-7112.
25. Doring V, Mootz HD, Nangle LA, Hendrickson TL, de Crecy-Lagard V, Schimmel P, Marliere P. Enlarging the amino acid set of escherichia coli by infiltration of the valine coding pathway. Science 2001; 292:501-504.
26. Wang L, Brock A, Herberich B, Schultz PG. Expanding the genetic code of escherichia coli. Science 2001; 292:498-500.
27. Thorson JS, Chapman E, Murphy EC, Schultz PG, Judice JK. Linear free energy analysis of hydrogen bonding in proteins. J. Am. Chem. Soc. 1995; 117:1157-1158.
28. Koh JT, Cornish VW, Schultz PG. An experimental approach to evaluating the role of backbone interactions in proteins using unnatural amino acid mutagenesis. Biochemistry 1997; 36:11314-11322.
29. Chapman E, Thorson JS, Schultz PG. Mutational analysis of backbone hydrogen bonds in staphylococcal nuclease. J. Am. Chem. Soc. 1997; 119:7151-7152.
30. Mendel D, Ellman JA, Chang ZY, Veenstra DL, Kollman PA, Schultz PG. Probing protein stability with unnatural amino acids. Science 1992; 256:1798-1802.
31. Bilgicer B, Xing X, Kumar K. Programmed self-sorting of coiled coils with leucine and hexafluoroleucine cores. J. Am. Chem. Soc. 2001; 123:11815-11816.
32. Lee HY, Lee KH, Al-Hashimi HM, Marsh EN. Modulating protein structure with fluorous amino acids: increased stability and native-like structure conferred on a 4-helix bundle protein by hexafluoroleucine. J. Am. Chem. Soc. 2006; 128:337-343.
33. Judice JK, Gamble TR, Murphy EC, de Vos AM, Schultz PG. Probing the mechanism of staphylococcal nuclease with unnatural amino acids: kinetic and structural studies. Science 1993; 261:1578-1581.
34. England PM, Zhang Y, Dougherty DA, Lester HA. Backbone mutations in transmembrane domains of a ligand-gated ion channel: implications for the mechanism of gating. Cell 1999; 96:89-98.
35. Dang H, England PM, Farivar SS, Dougherty DA, Lester HA. Probing the role of a conserved ml proline residue in 5-hydroxy-tryptamine(3) receptor gating. Mol. Pharmacol. 2000; 57:1114- 1122.
36. Lummis SC, Beene DL, Lee LW, Lester HA, Broadhurst RW, Dougherty DA. Cis-trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature 2005; 438: 248-252.
37. Ting AY, Shin I, Lucero C, Schultz PG. Energetic analysis of an engineered cation-pi interaction in staphylococcal nuclease. J. Am. Chem. Soc. 1998; 120:7135-7136.
38. Zhong WG, Gallivan JP, Zhang YN, Li LT, Lester HA, Dougherty DA. From ab initio quantum mechanics to molecular neurobiology: a cation-pi binding site in the nicotinic receptor. Proc. Natl. Acad. Sci. U. S. A. 1998; 95:12088-12093.
39. Lummis SC, Bean D, Harrison NJ, Lester HA, Dougherty DA. A cation-pi binding interaction with a tyrosine in the binding site of the gabac receptor. Chem. Biol. 2005; 12:993-997.
40. Sagle LB, Zimmermann J, Matsuda S, Dawson PE, Romesberg FE. Redox-coupled dynamics and folding in cytochrome c. J. Am. Chem. Soc. 2006; 128:7909-7915.
41. Schultz KC, Supekova L, Ryu Y, Xie J, Perera R, Schultz PG. A genetically encoded infrared probe. J. Am. Chem. Soc. 2006; 128:13984-13985.
42. Wang J, Xie J, Schultz PG. A genetically encoded fluorescent amino acid. J. Am. Chem. Soc. 2006; 128:8738-8739.
43. Wang L, Xie J, Deniz AA, Schultz PG. Unnatural amino acid mutagenesis of green fluorescent protein. J. Org. Chem. 2003; 681:174-176.
44. Bae JH, Rubini M, Jung G, Wiegand G, Seifert MHJ, Azim MK, Kim J-S, Zumbusch A, Holak TA, Moroder L, Huber R, Budisa N. Expansion of the genetic code enables design of a novel “Gold” Class of green fluorescent proteins. J. Mol. Biol. 2003; 328:1071-1081.
45. Tsien RY. The green fluorescent protein. Annu. Rev. Biochem. 1998; 67:509-544.
46. Chattoraj M, King BA, Bublitz GU, Boxer SG. Ultra-fast excited state dynamics in green fluorescent protein: multiple states and proton transfer. Proc. Natl. Acad. Sci. U. S. A. 1996; 93:8362- 8367.
47. Maiti A, Roy S. Switching DNA-binding specificity by unnatural amino acid substitution. Nucleic Acids Res. 2005; 33:5896-5903.
48. Schumacher TN, Mayr LM, Minor DLJr, Milhollen MA, Burgess MW, Kim PS. Identification of d-peptide ligands through mirrorimage phage display. Science 1996; 271:1854-1857.
49. Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. Inhibiting hiv-1 entry: discovery of d-peptide inhibitors that target the gp41 coiled-coil pocket. Cell 1999; 99:103-115.
50. Xie J. Adding unnatural amino acids to the genetic repertoire. Ph.D. Thesis. The Scripps Research Institute. 2006. pp. 46-66.
51. Cook SN, Jack WE, Xiong X, Danley LE, Ellman JA, Schultz PG, Noren CJ. Photochemically initiated protein splicing. Angew. Chem. Int. Ed. Engl. 1995; 34:1629-1630.
52. Miller JC, Silverman SK, England PM, Dougherty DA, Lester HA. Flash decaging of tyrosine sidechains in an ion channel. Neuron 1998; 20:619-624.
53. Deiters A, Groff D, Ryu Y, Xie J, Schultz PG. A genetically encoded photocaged tyrosine. Angew. Chem. Int. Ed. Engl. 2006; 45:2728-2731.
54. Wu N, Deiters A, Cropp TA, King D, Schultz PG. A genetically encoded photocaged amino acid. J. Am. Chem. Soc. 2004; 126:14306-14307.
55. England PM, Lester HA, Davidson N, Dougherty DA. Site-specific, photochemical proteolysis applied to ion channels in vivo. Proc. Natl. Acad. Sci. U. S. A. 1997; 94:11025-11030.
56. Bose M, Groff D, Xie J, Brustad E, Schultz PG. The incorporation of a photoisomerizable amino acid into proteins in e. Coli. J. Am. Chem. Soc. 2006; 128:388-389.
57. Gandhi CS, Isacoff EY. Shedding light on membrane proteins. Trends Neurosci. 2005; 28:472-479.
58. Banghart M, Borges K, Isacoff E, Trauner D, Kramer RH. Light-activated ion channels for remote control of neuronal firing. Nat. Neurosci. 2004; 7:1381-1386.
See Also
Amino Acids, Chemistry of
Chemical Ligation: Peptide Synthesis
Natural and Unnatural Amino Acids, Synthesis of
Proteins, Chemical Chemical Modification of Proteins
Proteins: Structure, Function and Stability