CHEMICAL BIOLOGY
Chemistry of Aminoacyl-tRNA Synthetases
Mir Hussain Nawaz and Susan A. Martinis, Department of Biochemistry, University of Illinois, Urbana-Champaign, Illinois
doi: 10.1002/9780470048672.wecb008
Aminoacyl-tRNA synthetases (aaRSs) compose a family of essential enzymes that attach amino acids covalently to tRNA molecules during protein synthesis. Some aaRSs possess a hydrolytic amino acid editing function to ensure the fidelity of protein synthesis. In addition, aminoacylation can occur by indirect pathways that rely on mischarged tRNA intermediates and enzymes other than aaRSs. Throughout evolution, structural and functional divergence of aaRSs has yielded diverse secondary roles. Likewise, aaRS-like proteins with either sequence or structural similarities to synthetases exhibit functions that may or may not be related to aminoacylation. Many of these diverse aaRSs and aaRS-like proteins have been capitalized on by the microbial world and by medical research as targets for therapeutic agents such as antibiotics.
Aminoacyl-tRNA synthetases (aaRSs) are critical components of the translation machinery for protein synthesis in every living cell (1). Each aaRS enzyme in this family links a single amino acid covalently to one or more tRNA isoacceptors to form charged tRNAs. Identity elements within the tRNAs serve as molecular determinants or antideterminants that aid in selection by cognate aaRSs (2). Some aaRSs also have an amino acid editing mechanism to clear their mistakes (3). The canonical aaRSs and aaRS-like proteins have functionally diverged to perform many other important roles in the cell (4, 5). Their versatility and adaptability have provided unique opportunities to develop biotechnology tools and to advance medical research.
Ubiquity of aaRS Structure and Function
The aaRSs are an ancient family of enzymes that have a lengthy and diverse evolutionary history. For their central role in protein synthesis, the aaRSs generate aminoacylated tRNAs, which are transferred to an elongation factor such as EF-Tu in bacteria for delivery to the ribosome. Some aaRSs have diverged functionally to perform other secondary roles that impact critical cellular activities (4). In addition, paralogs that bear sequence homology to aaRS domains participate in a wide array of activities in the cell (4, 5).
Aminoacylation of tRNAs
The aaRSs catalyze the covalent attachment of an amino acid to the universal 3'-adenosine at the terminus of tRNA (1). Aminoacylation consists of a two-step reaction mechanism. First, an amino acid is activated via ATP to form an aminoacyl adenylate intermediate. Second, the amino acid is transferred to the 3'-end of tRNA, releasing AMP (Fig. 1a). Generally, amino acid activation can occur in the absence of tRNAs. However, glutamyl- (GluRS), glutaminyl- (GlnRS), arginyl- (ArgRS) and lysyl-I (LysRS-I) tRNA synthetases require tRNA as a cofactor for the activation step.

Figure 1. Aminoacylation reaction. (a) The overall aminoacylation reaction is performed in two steps by the aaRSs. Two modes of amino acid editing can hydrolyze the mischarged tRNA product (posttransfer editing) or misactivated aminoacyl adenylate intermediate (pretransfer editing). (b) The first step of the ATP-dependent aminoacylation reaction activates amino acid to generate an aminoacyl adenylate intermediate. (c) In the second step, the activated amino acid is transferred to the tRNA molecule and AMP is released.
Representative X-ray crystal structures have been solved for each of the 20 canonical aaRSs (Table 1). This wealth of molecular information has provided a starting point to begin to unravel diverse paradigms that govern substrate recognition, the mechanism of aminoacylation, and alternative functions. In many cases, multiple cocrystal complexes with various substrates, analogs, and inhibitors are available. Fifteen aaRS X-ray crystal structures have been solved in complexes with their cognate tRNAs. Crystallization of individual aaRS domains has also provided insight into structure-function relationships of this diverse family of enzymes.
Table 1. Classification of aaRSs (6, 8) and their crystal structures


*These aaRSs have a hydrolytic editing pathway to clear misactivated and mischarged amino acids.
The aaRSs can be divided evenly into two classes based on the architectures of their catalytic domains: the presence of specific consensus sequences and their chemical properties (6). The catalytic core of Class I aaRSs is composed of a Rossmann dinucleotide binding fold that is marked by two signature consensus sequences: KMSKS and HIGH (Fig. 2). Class II aaRSs are typically dimers or tetramers and possess a more unique catalytic core that is made up of seven antiparallel β-strands flanked by α-helices. These enzymes have three consensus motifs (Fig. 2). Motif 1 [GФXXФxxPФФ] is at the dimer interface, whereas motif 2 [FRXE-H/RXXXFXXX(D/E)] and motif 3 [GФGФGФ(D/E)RФФФФФ] are part of the active site (Ф represents a hydrophobic amino acid). Each of the two distinct classes of aaRSs aminoacylates a set of amino acids with diverse chemical properties that would be important for protein function. Interestingly, LysRS is represented in both classes (7). Recently, two new aaRSs that activate O-phosphoserine and pyrrolysine have also been added to the Class II group (8, 9).

Figure 2. Mode of ATP binding to aaRSs. (a) Active site of GluRS enzyme bound to ATP (PDB 1 N75). The consensus sequences KISKR (KMSKS) and HVGT (HIGH) are highlighted on the protein. (b) Active site of GlyRS enzyme bound to ATP (PDB 1B76). The consensus motifs 1, 2, and 3 are highlighted on the protein. Class I and Class II aaRSs bind ATP in an extended and bent conformation, respectively. ATP is shown as dark spheres.
Both classes of enzymes catalyze the common aminoacylation reaction but via different mechanisms (1). Class I and Class II aaRSs bind ATP in an extended and bent conformation, respectively (Fig. 2). In addition, class I enzymes bind the tRNA acceptor stem from the minor groove side, which orients the 2'-hydroxyl group of the A76 ribose for attachment of the amino acid (Fig. 3). In contrast, Class II aaRSs aminoacylate the 3'-hydroxyl of the terminal adenosine, because the enzyme binds to tRNA via its major groove. Class II phenylalanyl-tRNA synthetase (PheRS), which charges amino acids onto the 2'-hydroxyl group of A76 of tRNAPhe, is the only known exception to this rule.
The aaRSs possess diverse polypeptide domains and insertions, in addition to their catalytic core. Likely, these domains evolved to enhance specificity and fidelity and, in some cases, confer other functions (4, 5). One such domain is the C-terminal anticodon-binding domain that is widely varied (1). For example, GluRS and GlnRS have highly conserved active sites within their canonical aminoacylation cores but have appended N-terminal anticodon-binding domains that are composed primarily of either α-helices or β-strands, respectively. In addition, common RNA-binding protein domains such as the OB-fold have been incorporated into aaRSs such as LysRS-II. In at least half of the aaRSs, an internal or appended domain confers amino acid editing activity (3).

Figure 3. Mode of tRNA binding to aaRSs. (a) Active site of GluRS enzyme bound to ATP and tRNAGlu (PDB 1 N77). (b) Active site of AspRS enzyme bound to Asp-AMP and tRNAAsp (PDB 1 C0A). Class I and Class II enzymes bind the tRNA acceptor stem from the minor and major groove sides, respectively. This orients either the 2'-OH or 3'-OH of A76 for specific attachment of the amino acid. ATP is represented by dark spheres, and tRNA is shown as a tube.
Alternative chemical activities
By virtue of their long evolutionary history, as well as of their capacity to bind RNA, ATP, and other small molecules such as amino acids, the aaRSs have been recruited to carry out many diverse alternative functions in cells (4) (Fig. 4a). One such function includes capitalizing on its aminoacylation function to proofread tRNA processing and maturation in the nucleus that occurs before tRNA export to the cytoplasm for protein synthesis. In addition, aminoacylation of tRNA-like structures, such as the 3'-end of viral genomes in plants and tmRNA in Escherichia coli, is important for viral replication and for ribosome recycling, respectively.
The RNA binding properties of aaRSs, such as leucyl- (LeuRS) and tyrosyl- (TyrRS) tRNA synthetases, have also been exploited to enable excision of self-splicing group I introns in some mitochondria. Others, including threonyl- (ThrRS) and alanyl- (AlaRS) tRNA synthetases from E. coli, are involved in transcriptional and translational regulation through interactions with their mRNA and DNA, respectively. PheRS also binds specifically to DNA, but the function of this property remains unclear. Some of the most diverse roles for aaRSs include cytokine and anti-angiogenic activities for TyrRS and tryptophanyl-tRNA synthetase (TrpRS), respectively.
aaRS-like proteins
Proteins with sequence homology to aaRSs perform diverse cellular functions (5) (Fig. 4b). Some of these proteins resulted from aaRS gene duplications and are called paralogs. Other paralogs are composed of just one domain of an aaRS that has multiple domains.

Figure 4. Cellular roles of aaRSs and aaRS-like proteins. (a) Alternative functions of aaRSs. The canonical aaRSs perform diverse functions in the cell that are distinct from their primary role of aminoacylation in protein synthesis 4. (b) AaRS-like proteins. Proteins that either resemble aaRSs or are paralogs of aaRSs are widespread in nature and carry out a variety of activities 5.
Paralog examples include E. coli YadB, which resembles GluRS. YadB attaches glutamate to a queuosine base yielding an essential tRNAAsp anticodon modification. In addition, two methionyl-tRNA synthetase (MetRS)-like proteins called Arc1p and Trbp111 bind to tRNA. Although the role of Trbp111 is not understood, Arc1p aids in delivery of tRNAs to their cognate aaRSs subsequent to nuclear export.
Several aaRSs possess amino acid editing domains, which hydrolytically clear mistakes in cis (3) (Table 1). Interestingly however, some archaeal and bacterial synthetases rely on paralogs of editing domains that hydrolyze mischarged products in trans (10, 11). These products include the YbaK and ProX (or PrdX), which edit Ala-tRNAPro, and AlaX that hydrolyzes mischarged Ser-tRNAAla and Gly-tRNAAla.
Several aaRS-like proteins are involved in metabolic pathways (1). For example, E. coli asparagine synthase, an aspartyl-tRNA synthetase (AspRS)-like enzyme, catalyzes the synthesis of asparagine from aspartate and ATP. A paralog of LysRS-II, called PoxA/GenX, is important for pyruvate oxidase activity in E. coli and Salmonella typhimurium and for virulence in S. typhimurium. The E. coli biotin synthetase/repressor protein (BirA), which has a domain that resembles structurally the seryl-tRNA synthetase (SerRS) catalytic domain, activates biotin to modify posttranslationally various metabolic proteins involved in carboxylation and decarboxylation. BirA can also bind DNA and regulate its own transcription using biotin as a corepressor. A histidyl-tRNA synthetase (HisRS)-like protein from Lactococcus lactis, HisZ is involved in the allosteric activation of the phosphoribosyl-transferase reaction.
Binding of uncharged tRNA to a HisRS-like domain of the GCN2 kinase protein in Saccharomyces cerevisiae under starvation and stress conditions elicits a cascade of molecular events to upregulate genes for amino acid and nucleotide biosynthesis. The accessory subunit Poly P of mtDNA polymerase y, which shares structural homology to both the catalytic and the anticodon binding domains of glycyl-tRNA synthetase (GlyRS), increases the processivity of the catalytic subunit of DNA-Poly and was proposed to serve as a primer recognition factor.
Chemistry of Aminoacylation
Recognition of tRNAs by aaRSs
One of the most significant features of the aminoacylation reaction is the specific recognition of a set of tRNA isoacceptors by their corresponding aaRS (2). Accurate tRNA:aaRS pairing relies on molecular “identity elements” that are composed of individual nucleotides, modifications, base pairs, or structural motifs. Positive identity elements (or determinants) enhance the selection of a tRNA by its cognate aaRS, whereas negative identity elements (or antideterminants) prevent the formation of incorrect tRNA:aaRS pairs. These identity elements can be classified as either major or minor elements based on the level of their impact on recognition.
Most identity elements are present at the two distal ends of the molecule: the acceptor stem and the triplet anticodon (Fig. 5). In the acceptor stem, the unpaired discriminator base N73 is a crucial recognition factor for most aaRSs. Also, the first few acceptor stem base pairs often serve as determinants for many tRNAs. The structural domains of aaRSs play specific roles in the recognition of tRNAs (12). In general, the more conserved catalytic domain binds the acceptor stem during aminoacylation. As indicated, diverged domains in most aaRSs interact directly with the tRNA anticodons to enable discrimination. The only exceptions are LeuRS, SerRS, and AlaRS, which do not require anticodon interactions for aminoacylation. SerRS and LeuRS interact, respectively, with up to four and six tRNA isoacceptors that have highly varied anticodons. In some aaRSs, accessory domains enhance recognition.
Unique architectural features of tRNAs also can serve as identity elements (2). For example, the long variable loop of tRNASer interacts specifically with SerRS. In addition, the tertiary G15:G48 Levitt base pair in E. coli tRNACys, and the triplet interaction in tRNAAsp, is formed between G45, and the G10:U25 pair confers identity. Occasionally, modified nucleotides can act as determinants, as in the case of E. coli tRNAIle, tRNAGlu, tRNALys, and yeast tRNAIle. All of these tRNAs contain modifications in the anticodon loop.
Antideterminants are defined as nucleotides that block mis- aminoacylation of the tRNA by a noncognate aaRS (2). A few examples of unmodified nucleotides or base pairs that act as antideterminants include A73 in human tRNALeu that hinders aminoacylation by SerRS, U34 in yeast tRNAIle that interferes with MetRS binding, and a G3:U70 base pair in yeast tRNAAla that blocks ThrRS. In addition, lysidine 34 and m1G37 are located in the anticodon structures of E. coli tRNAIle and yeast tRNAAsp and serve as antideterminants against MetRS and ArgRS enzymes, respectively. Interestingly, tRNAs that are charged by Class I aaRSs possess antideterminants against Class II aaRSs and vice versa (2).

Figure 5. Protein-RNA interactions of aaRSs. The cloverleaf secondary structure of tRNALeu folds into an L-shaped tertiary molecule. The tRNA can bind in an aminoacylation complex, where the 3' end is located in the canonical Class I or Class II core as shown in the upper right for the P. horikoshii LeuRS-tRNALeu aminoacylation complex. In aaRSs that edit, a second complex can be formed, where the 3' end interacts with a separate domain such as the connective polypeptide insertion (CP1) that contains a hydrolytic active site as shown in the lower right for the T. thermophilus LeuRS-tRNALeu editing complex. (Table (1); PDB files 1WZ2 and 2BYT).
Reaction mechanism
The overall two-step aminoacylation reaction relies on mechanistically distinct features of the Class I and Class II enzymes (1). In the Rossmann fold of Class I aaRSs, ATP binding is stabilized by interactions with the conserved KMSKS and HIGH consensus sequences. The β- and γ-phosphates interact with Mg2+. The α-NH3+ group of the bound amino acid is stabilized via hydrogen bonds with a conserved aspartate. Amino acid activation occurs by in-line nucleophilic displacement, where the a-carboxylate oxygen of the amino acid attacks the a-phosphorus atom of ATP (Fig. 1b). The flexible KMSKS peptide loop forms hydrogen bonds between the conserved lysine and serine and the pyrophosphate moiety of ATP during the transition state. Cleavage of the phosphoanhydride linkage between the α- and β-phosphates of ATP releases PPj. In the second step, the 2' ribose hydroxyl of the tRNA’s A76 attacks nucleophilically the carboxyl carbon of the aminoacyl adenylate intermediate to cleave the mixed anhydride. When amino acid is transferred to the tRNA molecule, AMP is released.
In Class II aaRSs, the bound ATP molecule interacts with motifs 2 and 3 (Fig. 2). The β- and γ-phosphates bend toward the adenine ring and are stabilized by three Mg2+ ions. The ribose of the ATP molecule adopts a 3'-endo conformation, as opposed to the 2'-endo conformation in Class I aaRSs. The amino acid side chain tends to bind by a lock-and-key mechanism, whereas its backbone interacts via an induced fit. Similar to Class I aaRSs, activation proceeds by an in-line nucleophilic displacement mechanism. The transition state is stabilized by interactions between the α- and γ-phosphates and arginine residues in motifs 2 and 3, respectively. The 3'-hydroxyl of the A76 ribose nucleophilically attacks the carboxyl carbon of the aminoacyl adenylate intermediate, forming aminoacyl-tRNA and AMP. The tRNA that is charged by either of the two classes is then transferred to an elongation factor such as EF-Tu in bacteria for delivery to the ribosome.
Amino acid editing
Nine aaRSs that include representatives of both classes possess a hydrolytic editing mechanism to clear misactivated amino acids (3) (Table 1). Hydrolysis of misactivated amino acids can occur either before or after transfer to tRNA (Fig. 1a). Pretransfer editing hydrolyzes adenylate intermediates but can be tRNA-dependent. Posttransfer editing cleaves mischarged tRNA. In general, aaRSs that edit seem to employ both pathways, although one may predominate.
Most aaRSs that possess an amino acid editing activity rely on a hydrolytic active site that has been integrated into their polypeptide chain. However, a few aaRSs are dependent on a mechanism where hydrolysis is carried out in trans by an independent domain (i.e., YbaK, ProX, and AlaX). MetRS and LysRS-II use the aminoacylation active site for editing and for clear mistakes by cyclizing the misactivated noncognate amino acid. Pretransfer editing has occurred both in an enzyme-based active site and in an aqueous environment via hydrolysis. The latter, which is commonly referred to as kinetic proofreading, relies on selective release of noncognate adenylates from the enzyme. These intermediates inherently are unstable and undergo hydrolysis in solution.
Most aaRSs that edit use a double-sieve mechanism. The aminoacylation active site serves as a coarse sieve and binds cognate amino acid as well as structurally related noncognate amino acids. A second hydrolytic active site acts as a fine sieve and hydrolyzes misactivated amino acids. The Class I aaRSs, LeuRS, isoleucyl- (IleRS), and valyl- (ValRS) tRNA synthetases rely on a homologous polypeptide insertion (connective polypeptide 1 or CP1 domain) for amino acid editing (Fig. 5). Class II aaRS editing domains are highly diverse. AlaRS, ThrRS, and prolyl-tRNA synthetase (ProRS) editing domains are homologous. However, the archaeal ThrRS domain is unrelated to its bacterial/eukaryotic counterpart or to the Class II homologs. Interestingly, PheRS has an editing domain that does not resemble that of any other aaRS.
Indirect pathways of aminoacylation
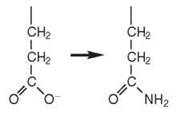
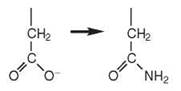
A complete set of 20 aaRSs to activate each of the 20 standard amino acids is not always present in every organism. These organisms rely on indirect mechanisms of aminoacy- lation that involve more than one enzyme and a mischarged tRNA intermediate (9, 12) (Table 2). For example, many bacteria, most archaea, and some eukaryotic organelles lack GlnRS and/or asparaginyl-tRNA synthetase (AsnRS) enzymes. Rather, misacylated tRNAs such as Glu-tRNAGln are produced by a nondiscriminating GluRS that interacts with both tRNAGlu and tRNAGln. A similar mechanism occurs for the formation of Asp-tRNAAsn. The mischarged tRNAs are then converted to the correct product by tRNA-dependent amidotransferases (AdTs). These AdTs include GatCAB, a heterotrimeric AdT found in bacteria, archaea, and eukaryotic organelles, which uses glutamine as an amide donor and amidates both Asp-tRNAAsn and Glu-tRNAGln. GatDE, a heterodimeric AdT found in archaea, amidates only Glu-tRNAGln.
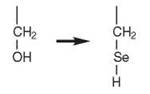
An indirect route of aminoacylation is also required for the incorporation of the so-called 21st amino acid selenocysteine (Sec) (9). In bacteria, Sec is inserted cotranslationally at an in-frame UGA codon that is upstream of an RNA stem-loop. In E. coli, SerRS produces the mischarged Ser-tRNASec, which is then converted to Sec-tRNASec by Sec synthase (SelA). A GTP-dependent elongation factor SelB binds to Sec-tRNASec and forms a complex that recognizes specific mRNA sequences called selenocysteine insertion elements (SECIS), that are located 3' to a UGA codon on a stalled ribosome-bound mRNA. It is also responsible for delivering Sec-tRNASec to the A site of the ribosome (9).
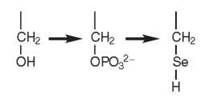
In archaea and eukaryotes, incorporation of Sec into polypeptides requires three steps involving three different enzymes. In the first step, SerRS mischarges tRNASec with serine to form Ser-tRNASec. Subsequently, a kinase, called O-phosphoseryl- tRNASec kinase (PSTK), phosphorylates serine to generate O-phosphoseryl-tRNASec (Sep-tRNASec). Finally, a pyridoxal phosphate-dependent enzyme called Sep-tRNA:Sec-tRNA synthase (SepSecS) (9) or Sec synthase (SecS) (13), converts Sep-tRNASec to Sec-tRNASec.
Many methanogenic archaeabacteria lack cysteinyl-tRNA synthetase (CysRS). Interestingly, a Class II enzyme called O-phosphoseryl-tRNA synthetase (SepRS) acylates tRNACys with O-phosphoserine (Sep) to form Sep-tRNACys, which is then converted to Cys-tRNACys by the enzyme Sep-tRNA:Cys-tRNA synthase (SepCysS). It has been proposed that this indirect pathway may be the sole route for cysteine biosynthesis in these organisms (9). The crystal structure of SepRS was recently solved (8). It is an ^synthetase whose quaternary structure resembles PheRS.
Pyrrolysine, the 22nd genetically encoded amino acid, is incorporated into a Methanosarcina barkeri protein in response to an in-frame UAG codon in the mRNA (9). Two different mechanisms have been proposed for the attachment of pyrrolysine to tRNAPyl. Pyrrolysyl-tRNA synthetase (PylRS) can aminoacylate tRNAPyl directly with pyrrolysine in a single step. In an indirect pathway, LysRS-I and LysRS-II, which belong to Class I and Class II aaRSs, respectively (Table 1), form a ternary complex with tRNAPyl and charge lysine onto it. The mischarged Lys-tRNAPyl would then be expected to be modified to Pyl-tRNAPyl by a mechanism that remains undefined (1).
Formylation of methionylated tRNAMet allows differentiation of the AUG start codon from internal AUG codons (14). MetRS aminoacylates tRNAfMet with methionine. A formyl group is linked covalently to the charged methionine via its amino moiety by the methionyl-tRNA formyltransferase enzyme, which uses N10-formyl tetrahydrofolate as the formyl donor. This fMet-tRNAfMet molecule binds directly to the P site of the ribosome to initiate protein synthesis, as compared with the A-site to which elongator tRNAs bind.
aaRSs: Expansion of the Genetic Code
The aaRS have enormous potential as tools to incorporate novel amino acids into proteins (15). Modified proteins that contain one or more nonstandard amino acids could confer unique chemical, physical, and/or biologic properties. These custom-designed proteins could be adapted as medicinal therapeutics and as biotechnology tools.
Incorporation of novel amino acids in proteins
Nonstandard amino acids can be either incorporated globally at multiple sites within a protein or inserted at specific locations (1, 15). Global misincorporation of nonstandard amino acids can produce protein polymers with altered physical properties that confer, for example, varied tensile strengths and elasticities (16). These unique biomaterials can be used in many medical applications, such as altering properties associated with cell adhesion. In other applications, routine replacement of methionine by selenomethionine aids in X-ray crystal structure determination.
Global incorporation of amino acid analogs into proteins has been achieved in vivo using E. coli strains that are auxotrophic for certain amino acids. Initially, cells are grown in the presence of the canonical amino acid until they reach sufficient biomass. The standard amino acid is then replaced by an analog in the media, followed by induction of a target protein for expression. The appropriate aaRS can also be overexpressed to boost charged tRNA production of analogs that are poorly activated.
Editing-defective aaRSs can also be capitalized on for global misincorporation of amino acids. For example, LeuRS has been used to incorporate oxonorvaline, a ketone-containing amino acid. These ketone groups, which are not found naturally in proteins, can be cross-linked readily to introduce diverse chemical properties to the protein. Other novel amino acids that have been incorporated successfully into proteins via editing defective aaRSs include α-aminobutyric acid by ValRS, norleucine and norvaline by IleRS and LeuRS, and unsaturated amino acids such as allylglycine, homoallylglycine, homopropargylgylcine, and 2-butynylalanine by LeuRS.
Table 2. Indirect pathways of aminoacylation (9, 12)
|
Product |
Mischarged intermediate(s) |
Organism |
Enzyme steps |
Chemical reaction |
|
Gln-tRNAGln |
Glu-tRNAGln |
Bacteria |
1. ND-GluRS1 |
|
|
|
|
Archaea Eukaryotic organelles |
2. Asp/GluAdT (GatCAB) or GluAdT (GatDE) |
|
|
Asn-tRNAAsn |
Asp-tRNAAsn |
Archaea Bacteria |
1. ND-AspRS1 |
|
|
|
|
2. Asp/GluAdT (GatCAB) |
||
|
Sec-tRNASec |
Ser-tRNASec |
Bacteria |
1. SerRS 2. SelA |
|
|
|
|
|
|
|
|
Sec-tRNASec |
Ser-tRNASec |
Archaea |
1. SerRS |
|
|
|
Sep-tRNASec |
Eukaryotes |
2. PSTK 3. SepSecS |
|
|
Cys-tRNACys |
Sep-tRNACys |
Archaea |
1. SepRS 2. SepCysS |
|
|
fMet-tRNAfMet |
Met-tRNAfMet |
Bacteria |
1. MetRS 2. Met-tRNA transfor-mylase |
|
|
|
|
|
|
|
|
*Pyl-tRNAPyl |
Lys-tRNAPyl |
Archaea |
1. LysRS-I:LysRS-II complex 2. unknown |
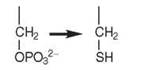
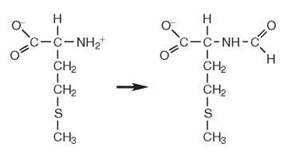
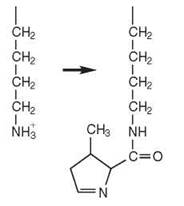
1 ND = nondiscriminating.
*An alternative direct pathway wherein pyrrolysine is charged to tRNAPyl by pyrrolysyl-tRNA synthetase (PylRS) has also been described (9).
Site-specific incorporation of novel amino acids in proteins (orthogonal aaRSs)
Site-specific insertion of novel amino acids in proteins originally was accomplished in vitro by generating mischarged suppressor tRNAs, which have anticodons that pair with amber “stop” codons. The tRNAs were aminoacylated chemically with natural and novel amino acids and were then incorporated at specific stop codons using in vitro translation systems.
Orthogonal aaRS:tRNA pairs were developed to incorporate nonstandard amino acids at specific protein sites in vivo (15). Within an organism, the orthogonal aaRS cannot recognize any endogenous tRNA molecules. Likewise, the orthogonal tRNA cannot be aminoacylated by any genome-encoded aaRSs.
Two general strategies have been employed to engineer successfully orthogonal pairs. First, aaRS and tRNA pairs from distant organisms; for example, S. cerevisiae were used in an E. coli system. Second, these aaRSs and tRNA molecules were altered via rational and/or random mutagenesis, typically us- ingin vivo selection systems, until they evolved to orthogonality.
Orthogonal pairs have been selected that can incorporate genetically more than 30 unnatural amino acids in E. coli, yeast, or mammalian cells. These amino acids include photocaged amino acids, glycosylated amino acids, and amino acids with chemically reactive groups that are used to modify selectively native proteins. This group also includes amino acids with novel properties that could be used as probes in protein structure-function analysis in several techniques, such as FRET, X-ray crystallography, NMR, and IR spectroscopies. Perhaps the best characterized is a Methanococcus janaschii TyrRS/tRNATyrCUA orthogonal pair that is expressed in E. coli and incorporates the synthetic amino acid O-methyl-L-tyrosine. Other examples include the yeast AspRS/tRNAAspCUA and yeast TyrRS/E. coli tRNAfMetCUA that function in E. coli , the LeuRS/tRNALeu5CUA pair that functions in yeast, E. coli TyrRS/tRNATyrCUA and GlnRS/tRNAfMetCUA pairs, the Bacillus subtilis TrpRS/tRNATrpCUA, and the E. coli TyrRS/ Bacillus stearothermophilus tRNATyrCUA pairs, all of which function in eukaryotic cells.
AARSs and Medicinal Therapeutics
Each member of the aaRS family is central to the viability of every living cell. Not only are they essential to protein synthesis, but also they are critical to many other important and diverse cellular activities (4). Species-specific inhibition of a single aaRS has been a proven drug target for antimicrobial development. Human aaRSs have long been associated with autoimmunity. More recent reports have linked aaRS defects to neurologic problems. Finally, the discovery of an impressive and growing list of aaRS alternative activities, which include roles in apoptosis, antiangiogenesis, and immunity provide new pathways to novel medicinal therapeutics.
aaRSs as antibiotic targets
Enzymes in the aaRS family are a promising target for the development of novel antibiotics (17). Selective inhibition of just one essential aaRS would be lethal to the pathogen. The best example is mupirocin, a commercially marketed IleRS inhibitor. Mupirocin, also known as pseudomonic acid, originally was isolated from Pseudomonas fluorescens and is used as a topical antibiotic against gram-positive bacteria, particularly Staphylococcus aureus. It binds directly to the first lysine of the conserved KMSKS sequence in the aminoacylation active site (18).
aaRS-linked autoimmune diseases
Antisynthetase syndrome is a condition that involves the production of antibodies that bind to and inhibit aaRSs (1). Several human aaRS antibodies have been identified as markers for this autoimmune disease. These antibodies are generally found in one third of all patients with polymyositis and dermatomyositis, which encompass chronic inflammatory muscle and skin disorders. These patients exhibit muscle inflammation, interstitial lung disease and arthritis, and mortality related to cardiopulmonary complications. To date, six human cytoplasmic aaRSs, including HisRS, ThrRS, AlaRS, IleRS, GlyRS, and AsnRS, have been identified as autoantigens and have been paired, respectively, with Jo-1, PL-7, PL-12, OJ, EJ, and KS autoantibodies. In any given condition, the patient’s serum has only one of the above autoantibodies. Although the origin of antisynthetase antibodies is not known, it is clear that elucidating their functions in autoimmune diseases may provide avenues to new therapeutics (1).
aaRS genetically linked neurologic diseases
Mutations in aaRS genes have been linked to progressive neurodegeneration and neuropathies. In one example, a single amino acid substitution (A734E) in the amino acid editing domain of mouse AlaRS enzyme results in the accumulation of misfolded proteins in purkinje cells within the adult cerebellum (19). This modest editing defect triggers the unfolded protein response and cell death in terminally differentiated neurons.
Charot-Marie-Tooth neuropathies can be caused by single mutations in genes encoding either TyrRS (20) or GlyRS (21, 22). The TyrRS-based mutation primarily affects aminoacylation and has been proposed to impair protein synthesis. In contrast, the molecular basis of pathogenesis remains unclear for GlyRS mutants, because they seem to retain aminoacylation activity (22).
aaRS-linked immunity and apoptosis
Under apoptotic conditions in human cells, TyrRS is secreted and then cleaved by the extracellular enzyme leukocyte elastase into two fragments. The two fragments have distinct cytokine activities (23). The C-terminal fragment is homologous to the endothelial monocyte-activating polypeptide-II cytokine. It stimulates the production of myeloperoxidase, TNF-a, tissue factor, and migration of polymorphonuclear and human umbilical vein endothelial cells. It also has leukocyte and monocyte chemotaxis activities, which are dependent on a heptapeptide located in the first β-strand of the β-barrel structure.
The N-terminal fragment (mini-TyrRS) is a proangiogenic factor. An ELR tripeptide found in CXC-chemokines with proangiogenic activity is located in the Rossmann fold of mini-TyrRS and is responsible for its angiogenic function. Mini-TyrRS also has interleukin-8-like activity, which includes stimulation of polymorphonuclear cell migration and binding to the IL-8 receptor.
aaRS anti-angiogenesis activity
Human TrpRS is involved in the inhibition of angiogenesis; the formation of capillaries from blood vessels (23). It is activated either by alternative splicing or by proteolysis. Alternative splicing results in six different mRNAs, one of which codes for a protein called mini-TrpRS. Mini-TrpRS is regulated by INF-y and blocks endothelial growth factor (EGF)-induced angiogenesis by initiating apoptosis of endothelial cells. The proangiogenic activity of mini-TyrRS and the antiangiogenic activity mini-TrpRS suggest that the two aaRSs might play cooperative, but opposing, roles in the regulation of angiogenesis.
Proteolysis by leukocyte elastase generates two N-terminally truncated fragments called T1-TrpRS and T2-TrpRS. The larger T1-TrpRS is similar in size to mini-TrpRS and has comparable angiostatic activity. T2-TrpRS, the smaller fragment, is an inhibitor of retinal and vascular EGF-induced angiogenesis and thus has tremendous potential as a novel anticancer agent. It targets specifically vascular-endothelial cadherin, a receptor expressed in endothelial cells and essential for normal vascular development. Deletion of a tRNA anticodon binding domain insertion, a helix-turn-helix motif of eight residues, abolished apoptotic activity (24).
References
1. Ibba M, Francklyn C, Cusack, S, eds. Aminoacyl-tRNA Synthetases. 2005. Landes Bioscience, Georgetown, TX.
2. Giege R, Sissler M, Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998; 26:5017-5035.
3. Hendrickson T, Schimmel P. Transfer RNA-dependent amino acid discrimination by aminoacyl-tRNA synthetases. In: Translation Mechanisms. Lapointe J, Brakier-Gingras L, eds. 2003. Kluwer Academic/Plenum Publishers, New York. p. 34-64.
4. Martinis SA, Plateau P, Cavarelli J, Florentz C. Aminoacyl-tRNA synthetases: a family of expanding functions. EMBO J. 1999; 18:4591-4596.
5. Schimmel P, Ribas De Pouplana L. Footprints of aminoacyl-tRNA synthetases are everywhere. Trends Biochem. Sci. 2000; 25:207- 209.
6. Ribas de Pouplana L, Schimmel P. Two classes of tRNA synthetases suggested by sterically compatible dockings on tRNA acceptor stem. Cell 2001; 104:191-193.
7. Ibba M, Morgan S, Curnow AW, Pridmore DR, Vothknecht UC, Gardner W, Lin W, Woese CR, and Soll D. A euryarchaeal lysyl-tRNA synthetase: resemblance to class I synthetases. Science 1997;278:1119-1122.
8. Kamtekar S, Hohn MJ, Park HS, Schnitzbauer M, Sauerwald A, Soll D, Steitz TA. Toward understanding phosphoseryl-tRNACys formation: the crystal structure of Methanococcus maripaludis phosphoseryl-tRNA synthetase. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:2620-2625.
9. Ambrogelly A, Palioura S, Soll D. Natural expansion of the genetic code. Nat. Chem. Biol. 2007; 3:29-35.
10. Ahel I, Korencic D, Ibba M, Soll D. Trans-editing of mischarged tRNAs. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:15422-15427.
11. An S, Musier-Forsyth K. Trans-editing of Cys-tRNAPro by Haemophilus influenzae YbaK protein. J. Biol. Chem. 2004; 279: 42359-42362.
12. Ibba M, Soll D. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000; 69:617-650.
13. Xu XM, Carlson BA, Mix H, Zhang Y, Saira K, Glass RS, Berry MJ, Gladyshev VN, Hatfield DL. Biosynthesis of selenocysteine on its tRNA in eukaryotes. PLoS Biol. 2006; 5:e4.
14. Raj Bhandary UL. Initiator transfer RNAs. J. Bacteriol. 1994;176: 547-552.
15. Wang L, Xie J, Schultz PG. Expanding the genetic code. Annu. Rev. Biophys. Biomol. Struct. 2006; 35:225-249.
16. Carrico IS, Maskarinec SA, Heilshorn SC, Mock ML, Liu JC, Nowatzki PJ, Franck C, Ravichandran G, Tirrell DA. Lithographic patterning of photoreactive cell-adhesive proteins. J. Am. Chem. Soc. 2007. In press.
17. Hurdle JG, O’Neill AJ, Chopra I. Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob. Agents Chemother. 2005; 49:4821-4833.
18. Silvian LF, Wang J, Steitz TA. Insights into editing from an ile-tRNA synthetase structure with tRNAile and mupirocin. Science 1999; 285:1074-1077.
19. Lee JW, Beebe K, Nangle LA, Jang J, Longo-Guess CM, Cook SA, Davisson MT, Sundberg JP, Schimmel P, Ackerman SL. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature 2006; 443:50-55.
20. Jordanova A, Irobi J, Thomas FP. Van Dijck P, Meerschaert K, Dewil M, Dierick I, Jacobs A, De Vriendt E, Guergueltcheva V, Rao CV, Tournev I, Gondim FA, D’Hooghe M, Van Gerwen V, Callaerts P, Van Den Bosch L, Timmermans JP, Robberecht W, Gettemans J, Thevelein JM, De Jonghe P, Kremensky I, Timmerman V. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet. 2006; 38:197-202.
21. Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin SQ, JordanovaA, Kremensky I, Christodoulou K, Middleton LT, Sivakumar K, Ionasescu V, Funalot B, Vance JM, Goldfarb LG, Fischbeck KH, Green ED. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet. 2003; 72:1293-1299.
22. Seburn KL, Nangle LA, Cox GA, Schimmel P, Burgess RW. An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model. Neuron 2006; 51:715-726.
23. Yang XL, Schimmel P, Ewalt KL. Relationship of two human tRNA synthetases used in cell signaling. Trends Biochem. Sci. 2004; 29:250-256.
24. Kise Y, Lee SW, Park SG, Fukai S, Sengoku T, Ishii R, Yokoyama S, Kim S, Nureki O. A short peptide insertion crucial for angiostatic activity of human tryptophanyl-tRNA synthetase. Nat. Struct. Mol. Biol. 2004; 11:149-156.
Further Reading
Allmang C, Krol A. Selenoprotein synthesis: UGA does not end the story. Biochimie 2006; 88:1561-1571.
Champagne KS, Sissler M, Larrabee Y, Doublie S, Francklyn CS. Activation of the hetero-octameric ATP phosphoribosyl transferase through subunit interface rearrangement by a tRNA synthetase paralog. J. Biol. Chem. 2005; 280:34096-34104.
Cusack S, Berthet-Colominas C, Hartlein M, Nassar N, Leberman R. A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-tRNA synthetase at 2.5 A. Nature 1990; 347:249-255.
Eriani G, Delarue M, Poch O, Gangloff J, Moras D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature 1990; 347:203-206.
Fan L, Sanschagrin PC, Kaguni LS, Kuhn LA. The accessory subunit of mtDNA polymerase shares structural homology with aminoacyl- tRNA synthetases: implications for a dual role as a primer recognition factor and processivity clamp. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:9527-9532.
Finn J, Tao J. Aminoacyl-tRNA synthetases as anti-infective drug targets. In Aminoacyl-tRNA Synthetases. Ibba M, Francklyn C, Cusack S, eds. 2005. Landes Bioscience, Georgetown, TX. p. 405-413.
First EA. Catalysis of the tRNA aminoacylation reaction. In: Aminoacyl-tRNA Synthetases. Ibba M, Francklyn C, Cusack S, eds. 2005. Landes Bioscience, Georgetown, TX. p. 328-363.
Francklyn C. tRNA synthetase-like proteins. In: Aminoacyl-tRNA Synthetases, Ibba M, Francklyn C, Cusack S, eds. 2005. Landes Bioscience, Georgetown, TX. p. 285-297.
Fukunaga R, Yokoyama S. Aminoacylation complex structures of leucyl-tRNA synthetase and tRNALeu reveal two modes of discriminator-base recognition. Nat. Struct. Mol. Biol. 2005; 12:915-922.
Geslain R, Ribas de Pouplana L. Regulation of RNA function by aminoacylation and editing? Trends Genet. 2004; 20:604-610.
Hendrickson TL, de Crecy-Lagard V, Schimmel P. Incorporation of nonnatural amino acids into proteins. Annu. Rev. Biochem. 2004; 73:147-176.
Ivakhno SS, Kornelyuk AI. Cytokine-like activities of some aminoacyl- tRNA synthetases and auxiliary p43 cofactor of aminoacylation reaction and their role in oncogenesis. Exp. Oncol. 2004; 26:250-255.
Kohrer C, RajBhandary UL. Proteins with one or more unnatural amino acids. In: Aminoacyl-tRNA Synthetases. Ibba M, Francklyn C, Cusack S, eds. 2005. Landes Bioscience, Georgetown, TX. p. 353-363.
Kron M, Hartlein M. Aminoacyl-tRNA synthetases and disease. In: Aminoacyl-tRNA Synthetases. Ibba M, Francklyn C, Cusack S, eds. 2005. Landes Bioscience, Georgetown, TX. p. 397-404.
Krzycki JA. The direct genetic encoding of pyrrolysine. Curr. Opin. Microbiol. 2005; 8:706-712.
Min B, Pelaschier JT, Graham DE, Tumbula-Hansen D, Still D. Transfer RNA-dependent amino acid biosynthesis: an essential route to asparagine formation. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:2678- 2683.
O’Donoghue P, Sethi A, Woese CR, Luthey-Schulten ZA. The evolutionary history of Cys-tRNACys formation. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:19003-19008.
Otani A, Slike BM, Dorrell MI, Hood J, Kinder K, Ewalt KL, Cheresh D, Schimmel P, Friedlander M. A fragment of human TrpRS as a potent antagonist of ocular angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:178-183.
Polycarpo C, Ambrogelly A, Berube A, Winbush SM, Mc Closkey JA, Crain PF, Wood JL, Still D. An aminoacyl-tRNA synthetase that specifically activates pyrrolysine. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:12450-12454.
Polycarpo C, Ambrogelly A, Ruan B, Tumbula-Hansen D, Ataide SF, Ishitani R, Yokoyama S, Nureki O, Ibba M, Soll D. Activation of the pyrrolysine suppressor tRNA requires formation of a ternary complex with class I and class II lysyl-tRNA synthetases. Mol Cell, 2003; 12:287-294.
Salazar JC, Ambrogelly A, Crain PF, Mc Closkey JA, Stoll D. A truncated aminoacyl-tRNA synthetase modifies RNA. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:7536-7541.
Sauerwald A, Zhu W, Major TA, Roy H, Palioura S, Jahn D, Whitman WB, Yates JR 3rd, Ibba M, Soll D. RNA-dependent cysteine biosynthesis in archaea. Science 2005; 307:1969-1972.
Sethi A, O’Donoghue P, Luthey-Schulten Z. Evolutionary profiles from the QR factorization of multiple sequence alignments. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:4045-4050.
Srinivasan G, James CM, Krzycki JA. Pyrrolysine encoded by UAG in Archaea: charging of a UAG-decoding specialized tRNA. Science 2002; 296:1459-1462.
Tzima E, Schimmel P. Inhibition of tumor angiogenesis by a natural fragment of a tRNA synthetase. Trends Biochem. Sci. 2006; 31:7-10.
Yuan J, Palioura S, Salazar JC, Su D, O’ Donoghue P, Hohn MJ, Cardoso AM, Whitman WB, Still D. RNA-dependent conversion of phosphoserine forms selenocysteine in eukaryotes and archaea. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:18923-18927.
See Also
Enzyme Catalysis, Chemistry of
Expanding the Genetic Code Through Chemical Biology
Protein Synthesis, Key Reactions of
Protein-Nucleic Acid Interactions
Translation: An Overview