Liquid-State Physical Chemistry: Fundamentals, Modeling, and Applications (2013)
Appendix C. The Lattice Gas Model
In this appendix we review briefly the lattice gas model [1], which is widely used in physical chemistry and known under different names in various fields, for example, as the regular solution model for liquid solutions, as the order–disorder model for alloys [2], or as the Ising model [3] for magnetic problems [4]. This broad range of applications explains its importance. For pure liquids the model was introduced by Frenkel [5] in 1932, and somewhat later independently by Eyring (and Cernuschi) [6]. It is one of the simplest models, if not the simplest model, showing a phase transition1), but we should note here that, although the model looks deceptively simple, its solution is complex.
C.1 The Lattice Gas Model
The basic idea of the lattice gas model is that the volume available to the fluid is divided into cells of molecular size. Usually, for simplicity, the cells are arranged in a regular lattice with coordination number z, for example, a simple cubic (SC) lattice with z = 6, a body-centered cubic (BCC) lattice with z = 8, or a face- centered cubic (FCC) lattice with z = 12. Normally, these cells are occupied by one molecule at most, representing repulsion, and only nearest-neighbor cell attractive interactions are considered.
The lattice gas model can be solved exactly for 1D, and in this case the model does not show a phase transition. It can also be solved exactly for 2D without an applied field, as was achieved for the first time by Onsager [7]; in this case, the model does show a phase transition. A complete exact solution (i.e., with applied field) is neither known for 2D, nor for 3D, so that one has to approximate. We will not deal with the exact 2D solution [2] but discuss here first the (conventional) zeroth approximation or mean field solution2) in terms of a model for liquid solutions, and thereafter the first approximation or quasi-chemical solution. In fact we have used these approximations in Chapters 8 and 11 already, although expressed in slightly different terms.
C.2 The Zeroth Approximation or Mean Field Solution
For a lattice model of a liquid solution we distribute molecules of type 1 and type 2 over the lattice (without any further holes). In Section 8.3, we showed that if we have N1 molecules of type 1 with N2 molecules of type 2, so that the total number becomes N = N1 + N2, we obtain the general relations
(C.1) ![]()
while the energy is given by
(C.2) ![]()
Here N11 (N22, N12) represent the number of type 1–1 (2–2, 1–2) pairs, ε11 (ε22, ε12) the energy of the pair 1−1 (2−2, 1−2), and the quantity w ≡ ε12 − ½ε11 − ½ε22 is denoted as interchange energy. To be consistent with Section 8.3 and Chapters 8 and 11, we use the notation zX ≡ N12, so that
(C.3) ![]()
The canonical partition function is easily constructed and we obtain
(C.4) ![]()
Recognizing that the term B ≡ ½(zε11N1 + zε22N2) is constant, this reduces to
(C.5) ![]()
where the sum Σ{X} is over all configurations having X type 1–2 pairs and qj is the internal partition function of molecule j. Equivalently, we write
(C.6) ![]()
where the sum ΣX is now over the number of configurations having X type 1–2 pairs with the degeneracy factor g(N1,N2,X) for each configuration labeled with (N1,N2,X).
In the zeroth approximation we consider that molecules are distributed randomly over the lattice, in spite of their interaction with others, which we restrict to their z nearest-neighbors. For the number of type 1–2 pairs zX we then get
(C.7) ![]()
(C.8) ![]()
The first expression was used in Chapter 8, but here we will use the second expression. The next step is to calculate g(N1,N2,X). Considering the distribution N1 molecules of type 1 and N2 of type 2 over N cells, one obtains for the number of configurations
(C.9) ![]()
Therefore, the partition function becomes
(C.10) ![]()
so that for the Helmholtz energy the result is
(C.11) ![]()
(C.12) ![]()
Consequently, the mixing Helmholtz energy reads
(C.13) ![]()
As a check, one can calculate the internal energy of mixing ΔmixU energy from ΔmixF via U = ∂(F/T)/∂(1/T) which leads, as expected, to
(C.14) ![]()
The coexistence line between a phase-separated system and a homogeneous system can be calculated from ∂ΔmixG/∂x2 = ∂ΔmixF/∂x2 = 0. This leads to
(C.15) ![]()
Using the transformation s = 2x2 − 1, the final expression is
(C.16) ![]()
The critical temperature Tcri is the temperature where the molecules start to order, and we determine Tcri by considering that tanh(½βzws) is a continuously increasing function of s passing through s = 0. Hence, there is always the solution s = 0 but for d[tanh(½βzws)]/ds|s=0 > 1, two other solutions occur (Figure C.1). Because this derivative equals ½βzw, we find Tcri = zw/2k. The composition as a function of T can be found by solving Eq. (C.16), either numerically or graphically, and the result is shown in Figure C.2a.
Figure C.1 The curves y = s and y = tanh(as) with (a) a = 0.5, (b) a = 1.0, and (c) a = 2.0, showing for the zeroth order solution, respectively, the homogeneous state (T > Tcri), the critical state (T = Tcri), and the phase-separated state (T < Tcri). All curves are plotted for s ≥ 0 only.
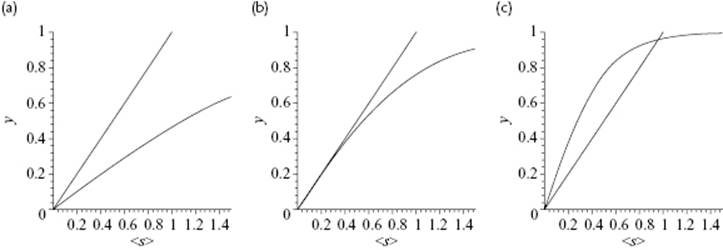
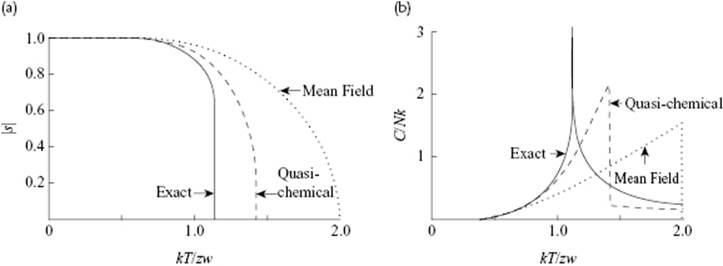
Figure C.2 The composition |s| = |2x2 − 1| where phase separation occurs (a) and the associated heat capacity C/Nk (b) as a function of kT/zw for the 2D square lattice, showing the zeroth order (mean field), the first order (quasi-chemical), and the exact solution.
The associated heat capacity CV can be found from CV = ∂U/∂T, with ![]() resulting in (after some calculation and plotted in Figure C.2b)
resulting in (after some calculation and plotted in Figure C.2b)
(C.17) ![]()
Note that CV shows a finite jump at T = Tcri with a maximum of 3Nk.
C.3 The First Approximation or Quasi-Chemical Solution
In the zeroth approximation, single molecules are distributed independently and randomly over the lattice. Improvement is possible by distributing randomly pairs of molecules instead of single molecules. This solution is known as the first approximation or quasi-chemical solution3).
C.3.1 Pair Distributions
Let us consider an independent distribution of pairs 1–1, 1–2, 2–1 and 2–2 over the lattice. Any such a distribution is characterized by the numbers N1, N2 and X, and this leads in the usual way to the number of possible configurations
(C.18) 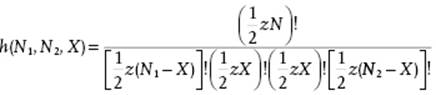
where we have used a separate entry for the 1–2 and 2–1 pairs in order to avoid the use of a symmetry number. This result cannot be correct, as the sum over all configurations zX should yield N!/N1!N2!. However, we can − at least approximately − remedy this defect by introducing a normalization factor c(N1,N2), and write for
(C.19) ![]()
(C.20) ![]()
We evaluate the sum G by taking its largest term (see Justification 5.2), and this term, labeled by X*, is obtained by setting ∂G/∂X = 0. The result is, after some straightforward manipulation,
(C.21) ![]()
This implies that the sum G for X*, labeled G*(N1,N2), becomes
(C.22) 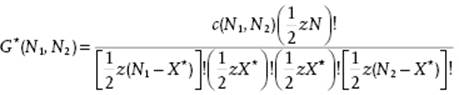
If we sum g(N1,N2,X) over all values of X, we must regain the total number of ways of placing N1 molecules of type 1 and N2 of type 2 over N = N1 + N2 sites. In other words, we also have G = N!/N1!N2! and we obtain for c(N1,N2)
(C.23) ![]()
Therefore, g(N1,N2,X) = N!h(N1,N2,X)/N1!N2!h(n1,n2,X*) or, in full
(C.24) 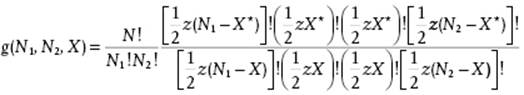
The configurational partition sum Q becomes
(C.25) ![]()
with, as usual, β = 1/kT. The maximum term trick is now played again and hence we seek the maximum term of Q, labeled ![]() , by setting ∂Q/∂X = 0, or equivalently, by setting ∂lnQ/∂X = 0. After a straightforward calculation we obtain
, by setting ∂Q/∂X = 0, or equivalently, by setting ∂lnQ/∂X = 0. After a straightforward calculation we obtain
(C.26) ![]()
For an explicit solution we define η2 ≡ exp(2βw) and the parameter α by
(C.27) ![]()
so that we can transform Eq. (C.26) to α2 − (1 − 4x1x2) = 4η2x1x2 (or α2 − (1 − 2x2)2 = 4η2x1x2), which has the solution α = [1 + 4x1x2(η2 − 1)]1/2 with xj = Nj/(N1 + N2). The energy of mixing becomes ΔmixU = 2x1x2zwN/(α + 1). Note that for α = 1 we regain the zeroth approximation.
C.3.2 The Helmholtz Energy
The configurational Helmholtz energy F is given by F = −kTlnQ and becomes
(C.28) 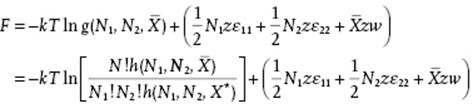
The evaluation of F is complex, and it is convenient to calculate first the chemical potential μj = ∂F/∂Nj. If this calculation is handled without thinking, it is a straightforward but tedious task. However, if we realize that ![]() is obtained from
is obtained from ![]() , the evaluation becomes straightforward. This implies that all terms resulting from
, the evaluation becomes straightforward. This implies that all terms resulting from ![]() taken together cancel. Moreover, X* is obtained from ∂lng/∂X = 0, implying that all terms from ∂lng/∂X* taken together also cancel. This renders the differentiation of F a relatively simple task, leading to
taken together cancel. Moreover, X* is obtained from ∂lng/∂X = 0, implying that all terms from ∂lng/∂X* taken together also cancel. This renders the differentiation of F a relatively simple task, leading to
(C.29) ![]()
To obtain μj − μj°, we realize that μj° is obtained from Eq. (C.28), again via μj = ∂F/∂Nj, but now with N2 = 0, leading to μj° = ½zβεjj. Writing F = N1μ1 + N2μ2 = N(x1μ1 + x2μ2), we obtain for the molar Helmholtz energy of mixing
(C.30) ![]()
Using the (absolute) activity λj = exp(βμj) and, as before, assuming that the activity can be replaced by the partial pressure, the activity coefficient γ becomes
(C.31) ![]()
In all these expressions X* and ![]() are given by Eq. (C.21) and (C.27), respectively.
are given by Eq. (C.21) and (C.27), respectively.
C.3.3 Critical Mixing
The above expression for ΔmixFm can be made more explicit by writing the various contributions in terms of α = [1 + 4x1x2(η2 − 1)]1/2 with η2 ≡ exp(2βw), x1 = N1/(N1 + N2) and x2 = N2/(N1 + N2). We find
(C.32) ![]()
(C.33) ![]()
(C.34) ![]()
so that ΔmixFm becomes
(C.35) ![]()
A similar substitution can be made for ![]() and Pj, leading to
and Pj, leading to
(C.36) 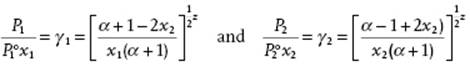
The critical point, due to symmetry, still occurs at x1 = x2 = ½. To obtain the expression for Tcri, we first derive the expression for the coexistence line, determined by ![]() . Upon substitution of the expression for
. Upon substitution of the expression for ![]() we obtain
we obtain
(C.37) ![]()
where the molecular ratio r ≡ x2/x1 and the abbreviation γ are introduced. Solving α yields α/(1 − 2x2) = (1 + γ)/(1 − γ), and thus leads to
(C.38) ![]()
Multiplying these two expressions leads to
(C.39) ![]()
Comparing with the transform of Eq. (C.26), α2 − (1 − 2x2)2 = 4η2x1x2, we obtain
(C.40) ![]()
Note that if r = r1 is a solution of Eq. (C.36), the other one is r2 = 1/r1.
The critical temperature Tcri is now obtained by putting r = 1 + δ in Eq. (C.40), expanding in powers of δ and taking the limit δ → 0. We thus obtain
(C.41) ![]()
For the SC lattice (z = 6) we have zw/kTcri = 2.433, while for the BCC lattice (z = 8) and FCC lattice (z = 12) we obtain zw/kTcri = 2.301 and zw/kTcri = 2.188, respectively. Note that if we take z = ∞, we regain the zeroth approximation zw/kTcri = 2.
Calculating, as before, the internal energy via U = ∂(F/T)/∂(1/T) is considerably more complex as for zeroth approximation and leads to
(C.42) ![]()
So far w was considered to be temperature independent but considering w as a temperature-dependent parameter presents no specific problems, and by using the definition u ≡ w − T(∂w/∂T), one can show that in this case one obtains ΔmixU = 2Nzux1x2/(α + 1).
C.4 Final Remarks
As stated in Section C.2, only a (partial) exact solution exists for 2D lattices. For a square lattice this solution reads zw/kTcri = 3.523, to be compared with zw/kTcri = 2.773 for the first and zw/kTcri = 2.00 for the zeroth approximation. The results from the exact solution for s and CV are shown, as are the results for the zeroth and first approximation, in Figure C.2. From these plots it becomes clear that considerable improvement is obtained by using the first approximation, but also that the exact solution is still far from being approached. Finally, the higher the number of dimensions, the better the mean field solution (see Chapter 16), which implies that the situation for 3D is somewhat better than for 2D.
Notes
1) For a very readable introduction to phase transitions, see Ref. [8].
2) Also denoted as the Bragg-Williams approximation.
3) Also denoted as the Bethe–Peierls approximation. For further details, see Refs [9] and [10].
References
1 Yang, C.N. and Lee, T.D. (1952) Phys. Rev., 87, 404 and 410.
2 Huang, K. (1987) Statistical Mechanics, 2nd edn, John Wiley & Sons, Inc., New York.
3 Ising, E. (1925) Z. Phys., 31, 253.
4 Brush, S.G. (1967) Rev. Mod. Phys., 39, 883.
5 Frenkel, J. (1946) Kinetic Theory of Liquids, Oxford University Press, Oxford. See also Dover Publishers reprint (1953).
6 (a) Eyring, H. (1936) J. Chem. Phys., 4, 283; (b) Cernuschi, F. and Eyring, H. (1939) J. Chem. Phys., 7, 547.
7 Onsager, L. (1944) Phys. Rev., 65, 117.
8 Stanley, H.E. (1971) Introduction to Phase Transitions and Critical Phenomena, Oxford University Press, New York.
9 Fowler, R.H. and Guggenheim, E.A. (1939) Statistical thermodynamics, Cambridge University Press.
10 Guggenheim, E.A. (1952) Mixtures, Clarendon, Oxford.