Liquid-State Physical Chemistry: Fundamentals, Modeling, and Applications (2013)
16. Some Special Topics: Phase Transitions
In Chapter 2 we reviewed general thermodynamics but avoided phase transitions. In this chapter, we discuss some aspects of these phenomena, dealing first with some general aspects and thereafter with discontinuous and continuous transitions, in particular the latter providing a rich part of physical chemistry.
16.1. Some General Considerations
By changing the conditions – such as P or T – for many materials, a transition from one phase to another can be induced. Under certain conditions even two phases of the same material may coexist. As each of the two phases has its own Gibbs energy expression, under these conditions the chemical potentials of the phases are equal. The Gibbs energy G itself is always continuous over the transition, but the partial derivatives ∂G/∂T and ∂G/∂P may be discontinuous (Figure 16.1). In that case, the phase transformation is denoted discontinuous1) (or first-order), while for the situation where the first derivative is continuous, but the higher derivatives are either zero or infinite, one speaks of a continuous (or second-order) phase transition.
Figure 16.1 Schematic of the behavior of the Gibbs energy G for two phases around (a) continuous phase transition and (b) discontinuous phase transformation. In both cases the stable states below the transition temperature have a Gibbs energy G1, while above the transition temperature the Gibbs energy is G2. The continuous transition occurs at the critical temperature Tcri with a continuous change in G, that is, ∂ΔG/∂T = 0, where ΔG = G2 − G1. The dotted line indicates the metastable continuation of the high temperature G below Tcri. The discontinuous transition occurs at a certain transition temperature Ttra with a discontinuous change in G (∂ΔG/∂T ≠ 0).

The angle of intersection of the G1 and G2 curve for phases 1 and 2, respectively, determines the entropy and volume change associated with the phase transformations, and hence the type of phase transition. Experimentally, it appears that by moving along the liquid–vapor (L-V) coexistence line over the critical point (CP), the differences in properties, in particular the density, between the liquid and gas phase vanish in a continuous way and the transition is continuous (Figure 16.2). Moving across the L-V curve from the liquid to the gas phase, and vice versa, leads to a discontinuous transition.
Figure 16.2 (a) Schematic of the phase equilibrium between the solid (S), liquid (L), and vapor (V) phase in the P–T plane, showing the triple point (TP) and critical point (CP). These are natural reference points as the melting temperature Tm and boiling temperature Tb depend on the environment, in particular the pressure P. For water, for example, Pcri = 218.3 atm, Tcri = 374.15 °C, ρcri = 320 kg m−3, and Ttri = 0.01 °C. While the transition over a coexistence line relates to a discontinuous phase transition, the transition over the critical point along the coexistence line relates to a continuous phase transition; (b) Schematic of the phase equilibrium in the P–V plane. The horizontal line indicates the equal area Maxwell construction. Above the CP only gases (G) can exist. The binodal line indicates the demarcation of global stability, while the spinodal line indicates the limits of local stability.
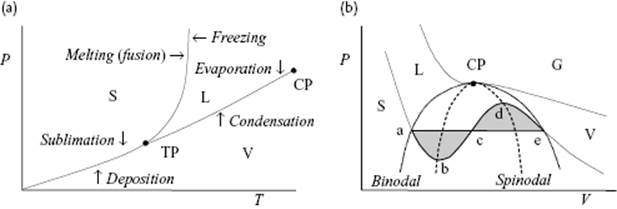
Following the coexistence (vapor pressure) line between liquid and vapor in the P–T diagram, we end at the critical point with temperature Tcri. In this process the density of the liquid decreases, while the density of the gas increases. At Tcri, the gas density ρgas and liquid density ρliq become identical. Moreover, for T < Tcri a meniscus – that is, a sharp transition region between liquid and vapor – is present except for temperatures close to Tcri (say within one degree), where the meniscus widens and suddenly disappears at Tcri. Figure 16.3 illustrates this behavior2).
Figure 16.3 The disappearing of the meniscus of benzene along the coexistence curve from far below (a) to close to (b) and just below (c) to just above Tcri (d).

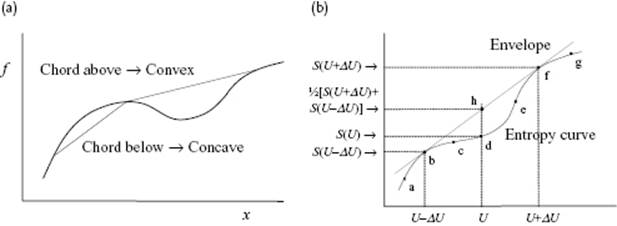
Before discussing some ideas on how to describe transitions, let us first discuss some stability considerations. For this we need the concept of convexity. A curve is convex (or convex up) if the chord is above the curve, whereas a curve is concave (or convex down) is the chord is below the curve (Figure 16.4a). Thermodynamics requires that for changes in entropy we always have (ΔS)U ≥ 0, which implies that entropy S is concave over its entire domain. If we obtain from a model or from experimental data a curve such as abcdefg in Figure 16.4b, the above implies that the envelope abhfg must be considered as the acting entropy function. Considering for the moment S as function energy U and volume V, concavity of S results in
(16.1) ![]()
where the reduction to the differential form only follows if ΔU → 0. Similarly,
(16.2) ![]()
where again the differential form only follows if ΔV → 0. In fact, these considerations also apply for a combined change of U and V,
(16.3) ![]()
leading again to Eq. (16.1) and (16.2) as well as to (Problem 16.3)
(16.4) ![]()
Figure 16.4 (a) Concave and convex interval of a function; (b) The relation to stability. If the curve abcdefg represents the entropy S, say as obtained from a model, as a function of energy U, the curve abhfg represents the entropy to be used since the entropy curve should be always concave. The range bcdef represents global instability (binodal), while the range cde (with c and e inflection points) represents local instability (spinodal).
So, the concavity leads to conditions for global stability while the differential forms lead to conditions for local stability. For example, Eq. (16.1) leads to
(16.5) ![]()
indicating that the molar heat capacity CV must be positive for a system to be thermally stable. Although Eq. (16.2) and (16.4) can also be used to establish further stability requirements, it is easier to employ thermodynamic potentials instead of the entropy. To that purpose, we first recall that the principle (ΔS)U ≥ 0 is always equivalent to (ΔU)S ≤ 0, stating that the energy U should always be convex. This leads to
(16.6) ![]()
(16.7) ![]()
which is fully analogous to the entropy case. It is convenient to consider also the Helmholtz and Gibbs energy. We first note that, if we have U(S,V) and F(T,V), then T = ∂U/∂S and S = −∂F/∂T, respectively. Therefore
(16.8) ![]()
Hence, the sign of ∂2F/∂T2 is the negative of the sign of ∂2U/∂S2, implying that if U is a convex function of S, then F is a concave function of T. This type of result holds for all transforms, so that we obtain for the Helmholtz function and Gibbs function, respectively,
(16.9) ![]()
Summarizing, the thermodynamic potentials – that is, the energy and its Legendre transforms – are convex functions of their extensive variables and concave functions of their intensive variables3).
From Eq. (16.9) we easily obtain
(16.10) ![]()
indicating that the isothermal compressibility κT must be positive for a system to be mechanically stable4). By combining CV ≥ 0 and κT ≥ 0 with κT − κS = TVα2/CP and CP − CV = TVα2/κT (see Chapter 2, Eq. 2.28), one can further infer that
(16.11) ![]()
The first of these equations states that, upon the addition of heat to a system, the temperature rises but more at constant volume than at constant pressure. The second equation states that, upon decreasing the volume of the system, the pressure increases, but more at constant temperature than at constant entropy.
Problem 16.1
Near the critical point the density profile of molecules with molar mass M over the meniscus is significantly influenced by gravity. If we describe the gravitational potential (approximately) by ϕ = gh, with g the acceleration due to gravity and h the height, the equilibrium condition reads d(μ + Mϕ) = 0.
a) What is the expression for the molar volume Vm?
b) Calculate the pressure P at height h with respect to the meniscus level h0.
c) Calculate the density ρ = M/Vm as a function of height h with respect to h0.
d) In many cases properties are scaled with respect to their value at critical point, for example, Tred = T/Tcri, Pred = P/Pcri and Vred = V/Vcri. What is the appropriate expression for μred in terms of Pcri, Vcri and Tcri?
e) Scaling the equilibrium condition d(μ + Mϕ) = d(μ + Mgh) = 0, we obtain dμred = −dhred. Calculate hcri in hred = h/hcri.
f) Consider Ne and H2O. For which compound is the width of the transition zone between liquid and gas near the critical point as characterized by hcri larger? What does the order of magnitude of hcri indicate to you? Use data as given in Appendix E.
Problem 16.2
Using the relation ∂2U/∂V2 ≥ 0, show that κS ≥ 0.
Problem 16.3
Show by expanding the left-hand side of Eq. (16.3) in a Taylor series to second order that:
![]()
where SUU ≡ ∂2S/∂U2, SUV ≡ ∂2S/∂U∂V, and SVV ≡ ∂2S/∂V2. Recalling that SUU ≤ 0, show that this expression can be written as
![]()
and that this subsequently leads to Eq. (16.4).