March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 17. Eliminations
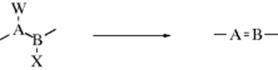
A so-called β-elimination reaction occurs when two groups are lost from adjacent atoms so that a new double1 (or triple) bond is formed. In general, the atom bearing a leaving group is the α, and the other the β atom. In an α-elimination, both groups are lost from the same atom to give a carbene (or a nitrene):
![]()
In a γ-elimination, a three-membered ring is formed:

Some of these processes were discussed in Chapter 10. Another type of elimination involves the expulsion of a fragment from within a chain or ring (X–Y–Z → X–Z + Y). Such reactions are called extrusion reactions. This chapter discusses β-elimination and extrusion reactions (see Sec. 2.F.vi); however, β-elimination in which both X and W are hydrogen atoms are oxidation reactions. They are treated in Chapter 19.
17.A. Mechanisms And Orientation
β-Elimination reactions may be divided into two types: one type taking place largely in solution, the other (pyrolytic eliminations) mostly in the gas phase. In the reactions, one group leaves with its electrons and the other without (i.e., it is pulled off), the latter most often being hydrogen. In these cases, the former leaves as the leaving group or nucleofuge. For pyrolytic eliminations, there are two principal mechanisms, one pericyclic and the other a free radical pathway. A few photochemical eliminations are also known (the most important is Norrish-type II cleavage of ketones, Sec. 7.A.vii), but these are not generally of synthetic importance2 and will not be discussed further. In most β-eliminations, the new bonds are C=C or C![]() C. The discussion of mechanisms is largely confined to these cases.3 Mechanisms in solution (E2, E1)4 and E1cB are discussed first. While standard methods are used to examine elimination reactions, new techniques (e.g., the velocity map ion imaging technique) have been used to study ultrafast elimination reactions.5
C. The discussion of mechanisms is largely confined to these cases.3 Mechanisms in solution (E2, E1)4 and E1cB are discussed first. While standard methods are used to examine elimination reactions, new techniques (e.g., the velocity map ion imaging technique) have been used to study ultrafast elimination reactions.5
17.A.i. The E2 Mechanism
In the E2 mechanism (elimination, bimolecular), the proton on the β carbon is pulled off by a base, leading to near-simultaneous expulsion of the leaving group (nucleofuge):
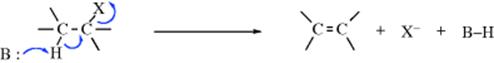
The mechanism takes place in one step and is kinetically second order: first order in substrate and first order in base. An ab initio study has produced a model for the E2 transition state geometry.6 The IUPAC designation is AxHDHDN, or more generally (to include cases where the electrofuge is not hydrogen), AnDEDN. It often competes with the SN2 mechanism (Sec. 10.A.i). With respect to the substrate, the difference between the two pathways is whether the species with the unshared pair attacks the carbon (and thus acts as a nucleophile) or the hydrogen (and thus acts as a base). As in the case of the SN2 mechanism, the leaving group may be positive or neutral and the base may be negatively charged or neutral.
Evidence for the existence of the E2 mechanism includes (1) the reaction displays the proper second-order kinetics; (2) when the hydrogen is replaced by deuterium in second-order eliminations, there is an isotope effect of from 3 to 8, consistent with breaking of this bond in the rate-determining step.7 However, neither of these results alone could prove an E2 mechanism, since both are compatible with other mechanisms also (e.g., see E1cB, Sec. 17.A.iii). The most compelling evidence for the E2 mechanism is found in stereochemical studies.8
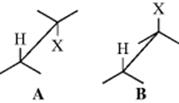
As will be illustrated in the examples below, the E2 mechanism is stereospecific: The five atoms involved (including the base) in the transition state must be in one plane. There are two ways for this to happen. The H and X may be trans to one another (A) with a dihedral angle of 180°, or they may be cis (B) with a dihedral angle of 0°.9 Conformation A is called antiperiplanar, and this type of elimination, in which H and X depart in opposite directions, is called anti elimination. Conformation B is syn periplanar, and this type of elimination, with H and X leaving in the same direction, is called syn elimination. Many examples of both kinds have been discovered. In the absence of special effects (discussed below), anti elimination is usually greatly favored over syn elimination, probably because A is a staggered conformation (Sec. 4.O.i) and the molecule requires less energy to reach this transition state than it does to reach the eclipsed transition state B. Solvent effects play an important role in the conformational preference. A few of the many known examples of predominant or exclusive anti elimination follow:
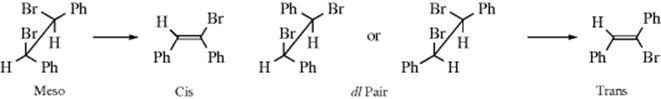
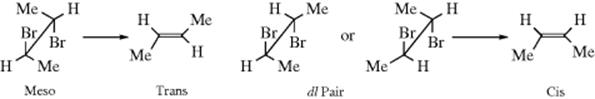
1. Elimination of HBr from meso-1,2-dibromo-1,2-diphenylethane gave cis-2-bromostilbene, while the (+) or (−) isomer gave the trans-alkene. This stereospecific result, which was obtained in 1904,10 demonstrates that in this case elimination is anti. Many similar examples have been discovered since. Obviously, this type of experiment need not be restricted to compounds that have a meso form. Anti elimination requires that an erythro dl pair (or either isomer) give the cis alkene, and the threo dl pair (or either isomer) give the trans isomer. This result has been found many times. Anti elimination has also been demonstrated in cases where the electrofuge is not hydrogen. In the reaction of 2,3-dibromobutane with iodide ion, the two bromines are removed (17-22). Using iodide as a base in this manner is unusual nowadays, and common bases are discussed in several sections below, including Reaction 17-13. In this case, the meso compound gave the trans alkene while the dl pair gave the cis:11
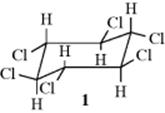
2. In open-chain compounds, rotation about C–C bonds usually lead to conformation in which H and X are antiperiplanar. However, in cyclic systems this is not always the case. There are nine stereoisomers of 1,2,3,4,5,6-hexachlorocyclohexane: seven meso forms and a dl pair (see Sec. 4.G). Four of the meso compounds and the dl pair) were treated with base to initiate elimination. Only one of these (1) has no Cl that is trans to an H. Of the other isomers, the fastest elimination rate was about three times as fast as the slowest, but the rate for 1 was 7000 times slower than that of the slowest of the other isomers.12 This result demonstrates that anti elimination is greatly favored over syn elimination, although the latter must be taking place on 1, but very slowly, to be sure.
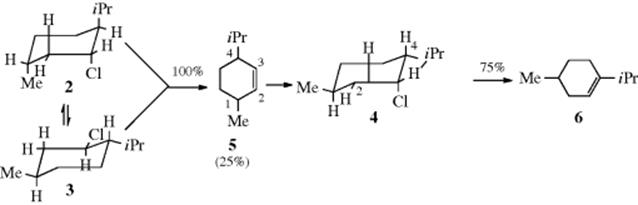
3. The preceding result shows that elimination of HCl in a six-membered ring proceeds best when the H and X are trans to each other. Adjacent trans groups on a six-membered ring can be diaxial or diequatorial (Sec. 4.O.ii) and the molecule is generally free to adopt either conformation, although one may have a higher energy than the other. Antiperiplanarity of the leaving group and the proton on the adjacent carbon requires that they be diaxial, even if this is the conformation of higher energy. The results with menthyl and neomenthyl chlorides are easily interpretable on this basis. Menthyl chloride has two chair conformations, 2 and 3. Compound 3, in which the three substituents are all equatorial, is the more stable and less reactive. The more stable chair conformation of neomenthyl chloride is 4, in which the chlorine is axial; there are axial hydrogen atoms on both C-2 and C-4. The results are the following: neomenthyl chloride gives rapid E2 elimination and the alkene produced is predominantly 6 (6/5 ratio is ~ 3:1) in accord with Zaitsev's rule (see Reaction 12-2, Sec. 17.B). Since an axial hydrogen is available on both sides, this factor does not control the direction of elimination and Zaitsev's rule is free to operate. However, for menthyl chloride, elimination is much slower and the product is entirely the anti-Zaitsev alkene 5. It is slow because the unfavorable conformation (2) must be achieved before elimination can take place. There is an axial hydrogen only on this side as the product must be 5.13
4. Anti elimination also occurs in the formation of triple bonds, as shown by elimination from cis- and trans-HO2C-CH=C(Cl)CO2H. In this case, the product in both cases is HO2CC![]() CCO2H, but the trans isomer reacts ~ 50 times faster than the cis compound.14
CCO2H, but the trans isomer reacts ~ 50 times faster than the cis compound.14
Some examples of syn elimination have been found in molecules where H and X could not achieve an antiperiplanar conformation.
1. The deuterated norbornyl bromide (7, X = Br) gave 94% of the product containing no deuterium.15 Similar results were obtained with other leaving groups and with bicyclo[2.2.2] compounds.16 In these cases the exo X group cannot achieve a dihedral angle of 180° with the endo β hydrogen because of the rigid structure of the molecule. The dihedral angle here is ~ 120°. Syn elimination with a dihedral angle of ~ 0° is clearly preferred to anti elimination where the angle is restricted to ~ 120°.
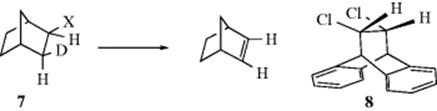
2. Molecule 8 is a particularly graphic example of the need for a planar transition state. In 8, each Cl has an adjacent hydrogen trans to it, and if planarity of leaving groups were not required, anti elimination could easily take place. However, the crowding of the rest of the molecule forces the dihedral angle to be ~ 120°, and elimination of HCl from 8 is much slower than from corresponding nonbridged compounds.17 Note that syn elimination from 8 is even less likely than anti elimination. Syn elimination can take place from the trans isomer of 8 (dihedral angle ~ 0°); this isomer reacted about eight times faster than 8.17
The examples given so far illustrate two points. (1) Anti elimination requires a dihedral angle of 180°. When this angle cannot be achieved, anti elimination is greatly slowed or prevented entirely. (2) For the simple systems so far discussed, syn elimination is not found to any significant extent unless anti elimination is greatly diminished by failure to achieve the 180° angle.
The concept of vinylogy was introduced in Section 6.B and in Reaction 10-68, category 4. Using this concept, a 1,2-elimination can be extended to give a 1,x-elimination when π bonds are incorporated between the carbon bearing the acidic proton and the leaving group (e.g., X-C-C=C-C, X-C-C=C-C=C-C or X-C-C![]() C-C).18
C-C).18
As noted in Section 4.Q.ii, six-membered rings are the only ones among rings of 4–13 members in which strain-free antiperiplanar conformations can be achieved. It is not surprising, therefore, that syn elimination is least common in six-membered rings. Cycloalkyltrimethylammonium hydroxides were subjected to elimination (Reaction 17-7) and the following percentages of syn elimination were found with each ring size: four-membered, 90%; five-membered, 46%; six-membered, 4% seven-membered, 31–37%.19 Note that the NMe3+ group has a greater tendency to syn elimination than do other common leaving groups (e.g., OTs, Cl, and Br).
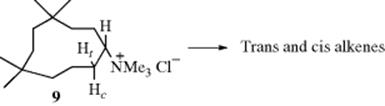
Other examples of syn elimination have been found in medium-ring compounds, where both cis and trans alkenes are possible (Sec. 4.K.i). As an illustration, elimination of 1,1,4,4-tetramethyl-7-cyclodecyltrimethylammonium chloride (9)20 gave mostly trans- but also some cis-tetramethylcyclodecenes as products. Note that trans-cyclodecenes, although stable, are less stable than the cis isomers. In order to determine the stereochemistry of the reaction, the elimination was repeated, this time using deuterated substrates. When 9 was deuterated in the trans position (Ht = D), there was a substantial isotope effect in the formation of both cis and trans alkenes, but when 9 was deuterated in the cis position (Hc = D), there was no isotope effect in the formation of either alkene. Since an isotope effect is expected for an E2 mechanism,21 these results indicated that only the trans hydrogen (Ht) was lost, whether the product was the cis or the trans isomer.22 This in turn means that the cis isomer must have been formed by anti elimination and the trans isomer by syn elimination. Anti elimination could take place from approximately the conformation shown, but for syn elimination the molecule must twist into a conformation in which the C–Ht and C–NMe3+ bonds are syn periplanar. Other types of evidence have also demonstrated this remarkable result, called the syn–anti dichotomy.23 The fact that syn elimination in this case predominates over anti (as indicated by the formation of trans-isomer in greater amounts than cis) has been explained by conformational factors.24 The syn–anti dichotomy has also been found in other medium-ring systems (8–12 membered),25 although the effect is greatest for 10-membered rings. With leaving groups,26 the extent of this behavior decreases in the order +NMe3 > OTs > Br > Cl, which parallels steric requirements. When the leaving group is uncharged, syn elimination is favored by strong bases and by weakly ionizing solvents.27
Syn elimination and the syn–anti dichotomy have also been found in open-chain systems, although to a lesser extent than in medium-ring compounds. For example, in the conversion of 3-hexyl-4-d-trimethylammonium ion to 3-hexene with potassium sec-butoxide, ~ 67% of the reaction followed the syn-anti dichotomy.28 In general, syn elimination in open-chain systems is only important in cases where certain types of steric effect are present. One such type is compounds in which substituents are found on both the β′ and the γ carbons (the unprimed letter refers to the branch in which the elimination takes place). The factors that cause these results are not completely understood, but the following conformational effects have been proposed as a partial explanation.29 The two anti- and two syn-periplanar conformations are, for a quaternary ammonium salt:
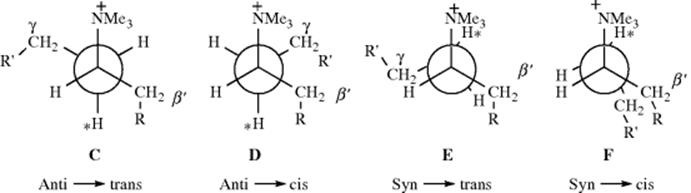
In order for an E2 mechanism to take place, a base must approach the proton marked ∗. In C, this proton is shielded on both sides by R and R′. In D, the shielding is on only one side. Therefore, when anti elimination does take place in such systems, it should give more cis product than trans. Also, when the normal anti elimination pathway is hindered sufficiently to allow the syn pathway to compete, the anti → trans route should be diminished more than the anti → cis route. When syn elimination begins to appear, it seems clear that E, which is less eclipsed than F, should be the favored pathway and syn elimination should generally give the trans-isomer. In general, deviations from the synanti dichotomy are greater on the trans side than on the cis. Thus, trans-alkenes are formed partly or mainly by syn elimination, but cis-alkenes are formed entirely by anti elimination. Predominant syn elimination has also been found in compounds of the form R1R2CHCHDNMe3+, where R1 and R2 are both bulky.30 In this case, the conformation leading to syn elimination (H) is also less strained than G, which gives anti elimination. The G compound has three bulky groups (including NMe3+) in the gauche position to each other.
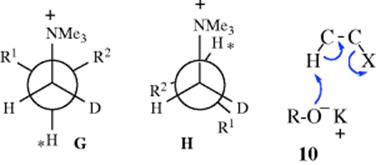
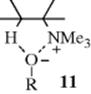
It was mentioned above that weakly ionizing solvents promote syn elimination when the leaving group is uncharged. This is probably caused by ion pairing, which is greatest in nonpolar solvents.31 Ion pairing can cause syn elimination with an uncharged leaving group by means of the transition state shown in 10. This effect was graphically illustrated by elimination from 1,1,4,4-tetramethyl-7-cyclodecyl bromide.32 The ratio of syn to anti elimination when this compound was treated with t-BuOK in the nonpolar benzene was 55.0. When the crown ether dicyclohexano-18-crown-6 was added (this compound selectively removes K+ from the t-BuO− K++ ion pair and thus leaves t-BuO− as a free ion), the syn/anti ratio decreased to 0.12. Large decreases in the syn/anti ratio on addition of the crown ether were also found with the corresponding tosylate and with other nonpolar solvents.33 However, with positively charged leaving groups the effect is reversed. Here, ion pairing increases the amount of anti elimination.34 In this case, a relatively free base (e.g., PhO−) can be attracted to the leaving group (see 11), putting it in a favorable position for attack on the syn β hydrogen, while ion pairing would reduce this attraction.
It can be concluded that anti elimination is generally favored in the E2 mechanism, but that steric (inability to form the antiperiplanar transition state), conformational, ion pairing, and other factors cause syn elimination to intervene (and even predominate) in some cases.
17.A.ii. The E1 Mechanism
The E1 mechanism is a two-step process in which the rate-determining step is ionization of the substrate to give a carbocation that rapidly loses a β proton to a base, usually the solvent:
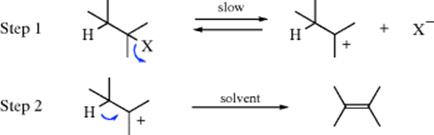
The IUPAC designation is DN + DE (or DN + DH). This mechanism normally operates without an added base. Just as the E2 mechanism competes with the SN2,35 so the E1 mechanism competes with the SN1. In fact, the first step of the E1 is exactly the same as that of the SN1 mechanism. The second step differs in that the solvent pulls a proton from the β carbon of the carbocation rather than attacking it at the positively charged carbon, as in the SN1 process. In a pure E1 reaction (without ion pairs, etc.), the product should be completely nonstereospecific, since bond rotation is possible in the carbocation before deprotonation.
Some of the evidence for the E1 mechanism is as follows:
1. The reaction exhibits first-order kinetics (in substrate) as expected. Of course, the solvent is not expected to appear in the rate equation, even if it were involved in the rate-determining step (Sec. 6.J.vi), but this point can be checked easily by adding a small amount of the conjugate base of the solvent. It is generally found that such an addition does not increase the rate of the reaction. If this more powerful base does not enter into the rate-determining step, it is unlikely that the solvent does. An example of an E1 mechanism with a rate-determining second step (proton transfer) has been reported.36
2. If the reaction is performed on two molecules that differ only in the leaving group (e.g., t-BuCl and t-BuSMe2+), the rates should obviously be different, since they depend on the ionizing ability of the molecule. However, once the carbocation is formed, if the solvent and the temperature are the same, it should suffer the same fate in both cases. This means that the nature of the leaving group does not affect the second step, and the ratio of elimination to substitution should be the same. The compounds mentioned in the example were solvolyzed at 65.3 °C in 80% aq ethanol with the following results:37
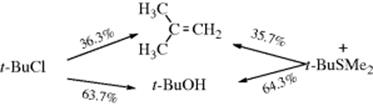
Although the rates were greatly different (as expected with such different leaving groups), the product ratios were the same, within 1%. If this had taken place by a second-order mechanism, the nucleophile would not be expected to have the same ratio of preference for attack at the β hydrogen compared to attack at a neutral chloride as for attack at the β hydrogen compared to attack at a positive SMe2 group.
3. Many reactions carried out under first-order conditions on systems where E2 elimination is anti proceed quite readily to give alkenes where a cis hydrogen must be removed, often in preference to the removal of a trans hydrogen. For example, menthyl chloride (2), which by the E2 mechanism gave only 5, under E1 conditions gave 68% 6 and 32% 5, since the steric nature of the hydrogen is no longer a factor here, and the more stable alkene (Zaitsev's rule, Reaction 12-2) is predominantly formed.
4. If carbocations are intermediates, rearrangements should occur with suitable substrates. These have often been found in elimination reactions performed under E1 conditions.
E1 reactions can involve ion pairs, just as is true for SN1 reactions (Sec. 10A.iii).38 This effect is naturally greatest for nondissociating solvents: It is least in water, greater in ethanol, and greater still in acetic acid. It has been proposed that the ion-pair mechanism (Sec. 10.A.iii, category 1) extends to elimination reactions too, and that the SN1, SN2, E1, and E2 mechanisms possess in common an ion-pair intermediate, at least occasionally.39
17.A.iii. The E1cB Mechanism40
In the E1 mechanism, X leaves first and then H is removed. In the E2 mechanism, H is removed, which triggers the expulsion of X. There is a third possibility: The H is removed first to form 12, and then X leaves. This reaction is a two-step process, called the E1cB mechanism,41 or the carbanion mechanism, since the intermediate is a carbanion, (12). The
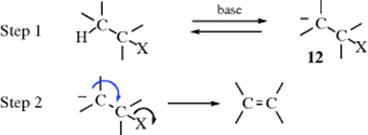
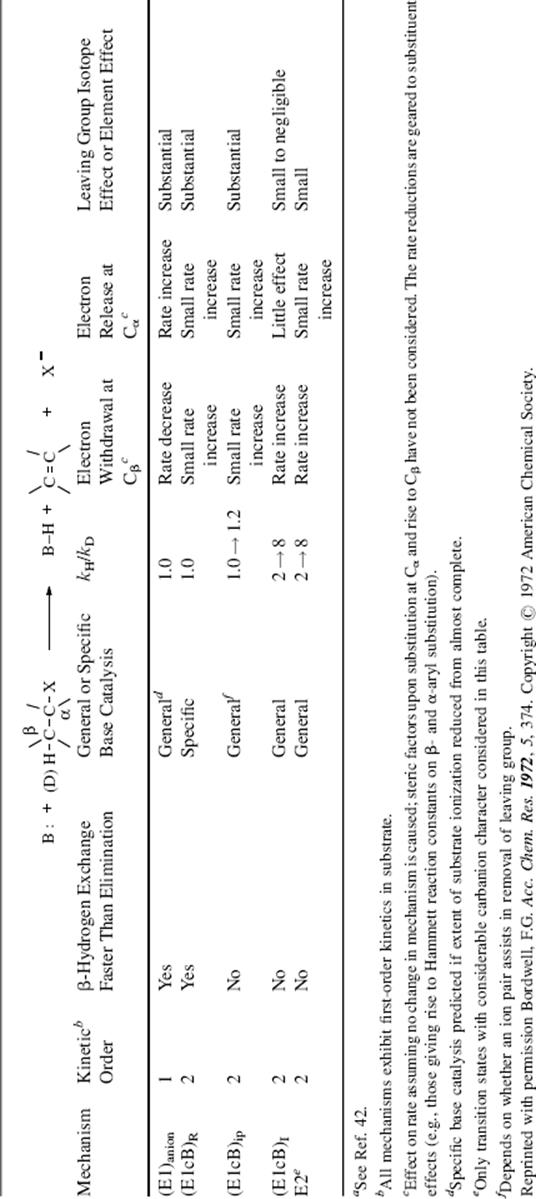
name E1cB comes from the fact that it is the conjugate base of the substrate that is giving up the leaving group (see the SN1cB mechanism, Sec. 10.G.iii, category 1). The IUPAC designation is AnDE + DN or AxhDH + DN (see Sec. 9.F). Three limiting cases can be distinguished: (1) The carbanion returns to starting material faster than it forms product: step 1 is reversible; step 2 is slow. (2) Step 1 is the slow step, and formation of product is faster than return of the carbanion to starting material. In this case, step 1 is essentially irreversible. (3) Step 1 is rapid, and the carbanion goes slowly to product. This case occurs only with the most stable carbanions. Here, too, step 1 is essentially irreversible. These cases have been given the designations: (1) (E1cB)R, (2) (E1cB)I (or E1cBirr), and (3) (E1)anion. Their characteristics are listed in Table 17.1.42 Investigations of the reaction order are generally not very useful (except for case 3, which is first order), because cases 1 and 2 are second order and thus difficult or impossible to distinguish from the E2 mechanism by this procedure.43 The greatest likelihood of finding the E1cB mechanism is expected in substrates that have (a) a poor nucleofuge and (b) an acidic hydrogen. In addition, most investigations have concerned such substrates. The following is some of the evidence in support of the E1cB mechanism:
Table 17.1 Kinetic Predictions for Base-Induced β-Eliminationsa
1. The first step of the (E1cB)R mechanism involves a reversible exchange of protons between the substrate and the base. In that case, if deuterium is present in the base, recovered starting material should contain deuterium. This was found to be the case in the treatment of Cl2C=CHCl with NaOD to give ClC![]() CCl. When the reaction was stopped before completion, there was deuterium in the recovered alkene.44 A similar result was found for pentahaloethanes.45 These substrates are relatively acidic. In both cases, the electron-withdrawing halogens increase the acidity of the hydrogen, and in the case of trichloroethylene there is the additional factor that a hydrogen on an sp2 carbon is more acidic than one on an sp3 carbon (Sec. 8.F, category 7). Thus, the E1cB mechanism is more likely to be found in eliminations yielding triple bonds than in those giving double bonds. Another likely place for the E1cB mechanism should be in reaction of a substrate like PhCH2CH2Br, since the carbanion is stabilized by resonance with the phenyl group. Nevertheless, no deuterium exchange was found here.46 If this type of evidence is a guide, then it may be inferred that the (E1cB)R mechanism is quite rare, at least for eliminations with common leaving groups (e.g., Br, Cl, or OTs), which yield C=C double bonds.
CCl. When the reaction was stopped before completion, there was deuterium in the recovered alkene.44 A similar result was found for pentahaloethanes.45 These substrates are relatively acidic. In both cases, the electron-withdrawing halogens increase the acidity of the hydrogen, and in the case of trichloroethylene there is the additional factor that a hydrogen on an sp2 carbon is more acidic than one on an sp3 carbon (Sec. 8.F, category 7). Thus, the E1cB mechanism is more likely to be found in eliminations yielding triple bonds than in those giving double bonds. Another likely place for the E1cB mechanism should be in reaction of a substrate like PhCH2CH2Br, since the carbanion is stabilized by resonance with the phenyl group. Nevertheless, no deuterium exchange was found here.46 If this type of evidence is a guide, then it may be inferred that the (E1cB)R mechanism is quite rare, at least for eliminations with common leaving groups (e.g., Br, Cl, or OTs), which yield C=C double bonds.
2. When the reaction shown was carried out in water containing acetohydroxamate buffers, a plot of the rate against the buffer concentration was curved and the rate leveled off at high buffer concentrations, indicating a change in rate-determining step.47 This rules out an E2 mechanism, which has only one step.48 When D2O was used instead of H2O as solvent, there was an initial inverse solvent isotope effect of 7.7 (the highest inverse solvent isotope effect yet reported).
![]()
That is, the reaction took place faster in D2O than in H2O. This is compatible only with an E1cB mechanism in which the proton-transfer step is not entirely rate determining. The isotope effect arises from a partitioning of the carbanion intermediate (12). This intermediate either can go to product or it can revert to starting compound, which requires taking a proton from the solvent. In D2O, the latter process is slower (because the O–D bond of D2O, cleaves less easily than the O–H bond of H2O), reducing the rate at which 12 returns to starting compound. With the return reaction competing less effectively, the rate of conversion of 12 to product is increased.
3. Substrates containing acidic hydrogen atoms and poor leaving groups are most likely to proceed by the E1cB mechanism. Compounds of the type ZCH2CH2OPh, where Z is an electron-withdrawing group (e.g., NO2, SMe2+, ArSO2, CN, CO2R), belong to this category, because OPh is a very poor leaving group (Sec. 10.A.iii, category 1). There is much evidence to show that the mechanism here is indeed E1cB.49 Isotope effects, measured for MeSOCD2CH2OPh and Me2S+CD2CH2OPh with NaOD in D2O, are ~ 0.7. This is compatible with an (E1cB)R mechanism, but not with an E2 mechanism for which an isotope effect of perhaps 5 might be expected (of course, an E1 mechanism is precluded by the extremely poor nucleofugal ability of OPh). The fact that kH/kD is less than the expected value of 1 is attributable to solvent and secondary isotope effects. Among other evidence for an E1cB mechanism in these systems is that changes in the identity of Z had a dramatic effect on the relative rates: a span of 1011 between NO2 and COO−. Note that elimination from substrates of the type RCOCH2CH2Y is the reverse of Michael-type addition to C=C bonds. Such addition involves initial attack by a nucleophile Y and subsequent protonation (see Sec. 15.A.ii). Thus the initial loss of a proton from substrates of this type (i.e., an E1cB mechanism) is in accord with the principle of microscopic reversibility.50 It may also be recalled that benzyne formation (Sec. 13.A.iii) can occur by such a process. It has been suggested that all base-initiated eliminations wherein the proton is activated by a strong electron-withdrawing group are E1cB reactions,51 but there is evidence that this is not the case when there is a good nucleofuge, the mechanism is E2 even when strong electron-withdrawing groups are present.52 On the other hand, Cl− has been found to be a leaving group in an E1cB reaction.53
Of the three cases of the E1cB mechanism, the one most difficult to distinguish from E2 is (E1cB)I. One way to make this distinction is to study the effect of a change in leaving group. This was done in the case of the three acenaphthylenes (13), where it was found that (1) the three rates were fairly similar, the largest being only about four times that of the smallest, and (2) in compound c (X = Cl, Y = F), the only product contained Cl and no F (i.e., only the poorer nucleofuge F departed
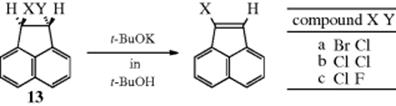
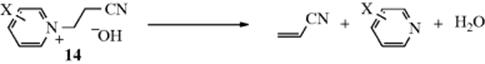
while Cl remained).54 Result (1) rules out all the E1cB mechanisms except (E1cB)I, because the others should all have considerable leaving group effects (Table 17.1). An ordinary E2 mechanism should also have a large leaving-group effect, but an E2 mechanism with substantial carbanionic character (see Section 17.A.iv) might not. However, no E2 mechanism can explain result (2), which can be explained by the fact that an α Cl is more effective than an α F in stabilizing the planar carbanion that remains when the proton is lost. Thus (as in the somewhat similar case of aromatic nucleophilic substitution, see Sec. 13.B.ii), when X− leaves in the second step, the one that leaves is not determined by which is the better nucleofuge, but by which has had its β hydrogen removed.55 Additional evidence for the existence of the (E1cB)I mechanism was the observation of a change in the rate-determining step in the elimination reaction of N-(2-cyanoethyl)pyridinium ions (14), treated with base, when X was changed.56 Once again, the demonstration that two steps are involved precludes the one-step E2 mechanism. Note that pyridyl systems appear to be a borderline case, and it is not obvious if the reaction involves a carbanion intermediate (E1cb, AxhDH + DN) or if the reaction proceeds by concerted loss of a proton and the halide (E2, ANDEDN) with attack by the base.57
4. An example of an (E1)anion mechanism has been found with the substrate 15, which when treated with methoxide ion undergoes elimination to 17, which is unstable under the reaction conditions and rearranges as shown.58Among the evidence for the proposed mechanism in this case were kinetic and isotope-effect results, as well as the spectral detection of 16.59
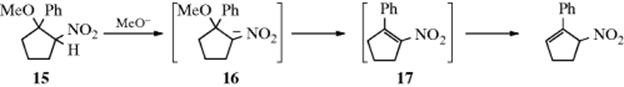
5. In many eliminations to form C=O and C![]() N bonds, the initial step is loss of a positive group (normally a proton) from the oxygen or nitrogen. These may also be regarded as E1cB processes.
N bonds, the initial step is loss of a positive group (normally a proton) from the oxygen or nitrogen. These may also be regarded as E1cB processes.
There is evidence that some E1cB mechanisms can involve carbanion ion pairs, for example,60
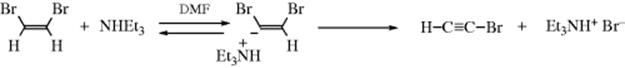
This case is designated (E1cB)ip; its characteristics are shown in Table 17.1.
17.A.iv. The E1–E2–E1cB Spectrum
In the three mechanisms so far considered, the similarities are greater than the differences. In each case, there is a leaving group that comes off with its pair of electrons and another group (usually hydrogen) that comes off without them. The only difference is in the order of the steps. It is now generally accepted that there is a spectrum of mechanisms ranging from one extreme, in which the leaving group departs well before the proton (pure E1), to the other extreme, in which the proton is removed first and then, after some time, the leaving group follows (pure E1cB). The pure E2 case would be somewhere in the middle, with both groups leaving simultaneously. However, most E2 reactions are not exactly in the middle, but somewhere to one side or the other. For example, the nucleofuge might depart just before the proton. This case may be described as an E2 reaction with a small amount of E1 character. The concept can be expressed by the question: In the transition state, which bond (C–H or C–X) has undergone more cleavage?61
Note that in both E1 and E2 reactions, removal of the hydrogen atom is an acid–base reaction, requiring a base. A stronger base is required for the E2, and a weaker base for E1. Further, the E1 reaction requires a solvent that facilitates ionization to a carbocation (e.g., aqueous media), whereas the E2 reaction is usually done in a protic solvent (e.g., an alcohol).
One way to determine just where a given reaction stands on the E1–E2–E1cB spectrum is to study isotope effects, which ought to tell something about the behavior of bonds in the transition state.62 For example, CH3CH2NMe3+showed a nitrogen isotope effect (k14/k15) of 1.017, while PhCH2CH2NMe3+ gave a corresponding value of 1.009.63 It would be expected that the phenyl group would move the reaction toward the E1cB side of the line, which means that for this compound the C–N bond is not as greatly broken in the transition state as it is for the unsubstituted one. The isotope effect bears this out, for it shows that in the phenyl compound, the mass of the nitrogen has less effect on the reaction rate than it does in the unsubstituted compound. Similar results have been obtained with SR2+ leaving groups by the use of ![]() isotope effects64 and with Cl (
isotope effects64 and with Cl (![]() ).65 The position of reactions along the spectrum has also been studied from the other side of the newly forming double bond by the use of H/D and H/T isotope effects,66 although interpretation of these results is clouded by the fact that β hydrogen isotope effects are expected to change smoothly from small to large to small again as the degree of transfer of the β hydrogen from the β carbon to the base increases67 (in Sec. 6.B, it was noted that isotope effects are greatest when the proton is half-transferred in the transition state), by the possibility of secondary isotope effects (e.g., the presence of a β deuterium or tritium may cause the leaving group to depart more slowly), and by the possibility of tunneling.68 Other isotope-effect studies have involved labeled α or β carbon, labeled α hydrogen, or labeled base.58
).65 The position of reactions along the spectrum has also been studied from the other side of the newly forming double bond by the use of H/D and H/T isotope effects,66 although interpretation of these results is clouded by the fact that β hydrogen isotope effects are expected to change smoothly from small to large to small again as the degree of transfer of the β hydrogen from the β carbon to the base increases67 (in Sec. 6.B, it was noted that isotope effects are greatest when the proton is half-transferred in the transition state), by the possibility of secondary isotope effects (e.g., the presence of a β deuterium or tritium may cause the leaving group to depart more slowly), and by the possibility of tunneling.68 Other isotope-effect studies have involved labeled α or β carbon, labeled α hydrogen, or labeled base.58
Another way to study the position of a given reaction on the spectrum involves the use of β aryl substitution. Since a positive Hammet ρ value is an indication of a negatively charged transition state, the ρ value for substituted β aryl groups should increase as a reaction moves from E1-to-E1cB-like along the spectrum. This has been shown to be the case in a number of studies;69 for example, ρ values of ArCH2CH2X increase as the leaving-group ability of X decreases. A typical set of ρ values was X = I, 2.07; Br, 2.14; Cl, 2.61; SMe2+, 2.75; F, 3.12.70 As seen previously, decreasing leaving-group ability correlates with increasing E1cB character.
Still another method measures volumes of activation.71 These are negative for E2 and positive for E1cB mechanisms. Measurement of the activation volume therefore provides a continuous scale for deciding just where a reaction lies on the spectrum.
17.A.v. The E2C Mechanism72
Certain alkyl halides and tosylates undergo E2 eliminations faster when treated with such weak bases as Cl− in polar aprotic solvents or PhS− than with the usual E2 strong bases (e.g., RO− in ROH).73 In order to explain these results, it was proposed74 that there is a spectrum75 of E2 transition states in which the base can interact in the transition state with the α carbon, as well as with the β hydrogen. At one end of this spectrum is a mechanism (called E2C) in which, in the transition state, the base interacts mainly with the carbon. The E2C mechanism is characterized by strong nucleophiles that are weak bases. At the other extreme is the normal E2 mechanism, here called E2H to distinguish it from E2C, characterized by strong bases. Transition state 18 represents a transition state between these extremes. Additional evidence76 for the E2C mechanism is derived from Br![]() nsted equation considerations (Sec. 8.D), from substrate effects, from isotope effects, and from the effects of solvents on rates.
nsted equation considerations (Sec. 8.D), from substrate effects, from isotope effects, and from the effects of solvents on rates.
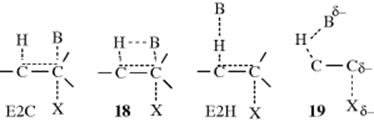
However, the E2C mechanism has been criticized, and it has been contended that all the experimental results can be explained by the normal E2 mechanism.77 McLennan and Lim78 suggested that the transition state is that shown as 19. An ion-pair mechanism has also been proposed.79 Although the actual mechanisms involved may be a matter of controversy, there is no doubt that a class of elimination reactions exists that is characterized by second-order attack by weak bases.80 These reactions also have the following general characteristics:81 (1) they are favored by good leaving groups; (2) they are favored by polar aprotic solvents; (3) the reactivity order is tertiary > secondary > primary, the opposite of the normal E2 order (Sec. 17.D.i); (4) the elimination is always anti (syn elimination is not found), but in cyclohexyl systems, a diequatorial anti elimination is about as favorable as a diaxial anti elimination (unlike the normal E2 reaction, Sec. 17.A.i, categories 2,3); (5) they follow Zaitsev's rule (see below), where this does not conflict with the requirement for anti elimination.