March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part I. Introduction
Chapter 5. Carbocations, Carbanions, Free Radicals, Carbenes, and Nitrenes
5.B. Carbanions
5.B.i. Stability and Structure105
Formally, a carbanion is a trivalent carbon atom with an unshared electron pair, and a formal charge of −1. In fact, there are few carbanions that do not have an anion-stabilizing group attached to the carbon atom. Stabilization may be by resonance delocalization or by orbital participation of an atom with d orbitals or orbitals associated withy a metal.
By definition, every carbanion possesses an unshared pair of electrons and is formally a base. When a carbanion donates an electron to a proton, it is converted to its conjugate acid (an acid–base reaction, see Chapter 8). If the carbanion (R3C:−) were available, reaction with an acid generates the conjugate acid (R3C–H), an alkane. The stability of the carbanion is directly related to the strength of the conjugate acid. The weaker that conjugate acid, the greater the base strength of the carbanion, and the lower the stability of the carbanion.106 Stability here is judged by diminished reactivity (lower electron-donating ability) with a proton. The greater the stability, the lower the electron-donating ability (lower reactivity) for reaction of the carbanion with a proton (any acid that is sufficiently strong), and hence the longer lived the carbanion. Thus the determination of the order of stability of a series of carbanions is equivalent to a determination of the inverse order of strengths of the conjugate acids, and one can obtain information about relative carbanion stability from a table of acid strengths (e.g., Table 8.1).
While formation of simple carbanions (e.g, CH3−) is rare, formation of a carbon–metal bond often generates a molecule (e.g., R3C–M, where M = a metal atom) that has a polarized bond in which the carbon is electron rich (δ−). An organic molecule that contains a carbon–metal bond is called an organometallic compound. Organometallic compounds where the metal is Mg, Li, or other metals are carbanion surrogates, and in much of their chemistry they react as if they were carbanions (see Reactions 12-22–12.39). Many such compounds are known, and organometallic chemistry is a very large area, occupying a borderline region between organic and inorganic chemistry. This section will discuss carbanions with little reference to a metal. Section 5.B.ii will discuss the structures of organometallic compounds, which are often carbanion surrogates.
Carbanions are very strong bases, and the conjugate acids of simple unsubstituted carbanions are very weak acids, with very few exceptions. Unfortunately, it is not easy to measure acid strengths of very weak acids. There is little doubt that carbanions are very unstable in solution, and in contrast to the situation with carbocations, efforts to prepare solutions in which carbanions (e.g., ethyl or isopropyl) exist in a relatively free state have not yet been successful. It has also not been possible to form these carbanions in the gas phase. Indeed, there is evidence that simple carbanions (e.g., ethyl and isopropyl) are unstable, losing an electron, which converts them to radicals.107Nevertheless, there have been several approaches to the problem. Applequist and O'Brien108 studied the position of equilibrium for the reaction:
![]()
This reaction was done in ether or an ether–pentane mixture. The reasoning in these experiments was that the R group that forms the more stable carbanion would be more likely to be bonded to lithium than to iodine. Carbanion stability was found to be in the order: vinyl > phenyl > cyclopropyl > ethyl > n-propyl > isobutyl > neopentyl > cyclobutyl > cyclopentyl. In a somewhat similar approach, Dessy et al.109 treated a number of alkylmagnesium compounds with a number of alkylmercury compounds in THF, setting up the equilibrium:
![]()
where the group of greater carbanion stability is linked to magnesium. The carbanion stability determined this way was phenyl > vinyl > cyclopropyl > methyl > ethyl > isopropyl. The two stability orders are in fairly good agreement, and they show that stability of simple carbanions decreases in the order methyl > primary > secondary. It was not possible to determine the position of tert-butyl by the experiments reported by Dessy et al.109, but there seems little doubt that it is still less stable. This stability order can be interpreted as solely a consequence of the field effect since resonance is absent. The electron-donating alkyl groups of isopropyl result in a greater negative charge density at the central carbon atom (compared with methyl), thus decreasing its stability. The results of Applequist and O'Brien108 show that β branching also decreases carbanion stability. Cyclopropyl occupies an apparently anomalous position, but this is probably due to the large amount of s character in the carbanionic carbon (see Sec. 5.B.i, category 2). Strongly electron-withdrawing groups (e.g., trifluoromethylsulfonyl) provide exceptional stability to carbanions.110
A different approach to the problem of hydrocarbon acidity, and hence carbanion stability, is that of Shatenshtein and Shapiro, who treated hydrocarbons with deuterated potassium amide and measured the rates of hydrogen exchange.111 The experiments did not measure thermodynamic acidity, since rates were measured, not positions of equilibria. They measured kinetic acidity; that is, which compounds gave up protons most rapidly (see Sec. 6.F for the distinction between thermodynamic and kinetic control of product). Measurements of rates of hydrogen exchange enable one to compare acidities of a series of acids against a given base even where the positions of the equilibria cannot be measured because they lie too far to the side of the starting materials; that is, where the acids are too weak to be converted to their conjugate bases in measurable amounts. Although the correlation between thermodynamic and kinetic acidity is far from perfect,112 the results of the rate measurements, too, indicated that the order of carbanion stability is methyl > primary > secondary > tertiary.111
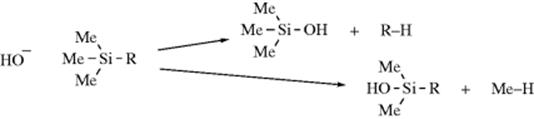
Experiments described above were done in solution, and experiments in the gas phase gave different results. In reactions of −OH with alkyltrimethylsilanes, it is possible to cleave either R or Me. Since the R or Me come off as a carbanion or incipient carbanion, the product ratio RH/MeH can be used to establish the relative stabilities of various R groups. From these experiments a stability order of neopentyl > cyclopropyl > tert-butyl > n-propyl > methyl > isopropyl > ethyl was found.113 On the other hand, in a different kind of gas-phase experiment, Graul and Squires114 were able to observe CH3− ions, but not the ethyl, isopropyl, or tert-butyl ions.
As mentioned above, carbanion-stabilizing groups can increase the stability of carbanions, which influences their ease of formation. Six structural features that lead to improved stability are listed:
1. Conjugation of the Unshared Pair with an Unsaturated Bond

In cases where a double or triple bond is located α to the carbanionic carbon, the ion is stabilized by resonance in which the unshared pair overlaps with the π electrons of the double bond. This factor is responsible for the stability of the allylic115 and benzylic116 types of carbanions:
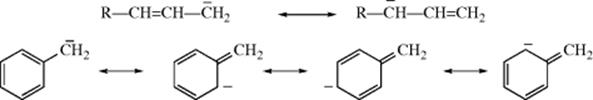
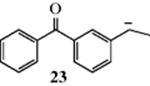
Diphenylmethyl and triphenylmethyl anions are still more stable due to extensive delocalization into the benzene rings, and can be kept in solution indefinitely if water is rigidly excluded.117 X-ray crystallographic structures have been obtained for Ph2CH− and Ph3C− enclosed in crown ethers.118 Carbanion 23 has a lifetime of several minutes (hours in a freezer at −20 °C) in dry THF.119 Condensed aromatic rings fused to a cyclopentadienyl anion are known to stabilize the carbanion.120
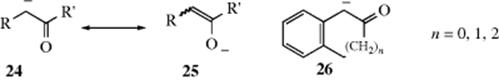
Where the carbanionic carbon is conjugated with a carbon–oxygen or carbon–nitrogen multiple bond (Y = O or N), the stability of the ion is greater than that of the triarylmethyl anions, since these electronegative atoms are more capable of bearing a negative charge than carbon. However, it is questionable whether ions of this type should be called a carbanion at all, since in the case of enolate ions, for example, 25 contributes more to the hybrid than 24 although such ions react more often at the carbon than at the oxygen. In benzylic enolate anions (e.g., 26), the conformation of the enolate can be coplanar with the aromatic ring or bent out of plane if the strain is too great.121 Enolate ions can also be maintained in solution in many cases, at least for minutes or hours at lower temperatures. In the case of carbanions at a carbon α to a nitrile, the “enolate” resonance form would be a ketene imine nitranion, but the existence of this species has been called into question.122 A nitro group is particularly effective in stabilizing a negative charge on an adjacent carbon, and the anions of simple nitro alkanes can exist in water. Thus the pKa for nitromethane is 10.2. Dinitromethane is even more acidic (pKa = 3.6). In contrast to the stability of cyclopropylmethyl cations (Sec. 5.A.ii), the cyclopropyl group exerts only a weak stabilizing effect on an adjacent carbanionic carbon.123
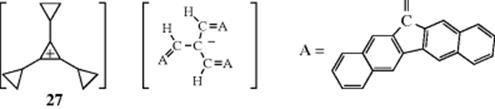
By combining a very stable carbanion with a very stable carbocation, Okamoto et al.124 were able to isolate the salt (27), as well as several similar salts, as stable solids. These are salts that consist entirely of carbon and hydrogen atoms.
2. Carbanions Increase in Stability with an Increase in the Amount of s Character at the Carbanionic Carbon. Thus the order of stability is
![]()
Acetylene, where the carbon is sp hybridized with 50% s character, is much more acidic than ethylene125 (sp2, 33% s), which in turn is more acidic than ethane, with 25% s character. Increased s character means that the electrons are closer to the nucleus, and hence of lower energy. As previously mentioned, cyclopropyl carbanions are more stable than methyl, owing to the larger amount of s character as a result of strain (see Sec. 4.Q.i).
3. Stabilization by Sulfur126 or Phosphorus. Attachment to the carbanionic carbon of a sulfur or phosphorus atom causes an increase in carbanion stability, although the reasons for this are in dispute. One theory is that there is overlap of the unshared pair with an empty d orbital127 (pπ–dπ bonding, see Sec. 2.H). For example, a carbanion containing the SO2R group would be written as follows:
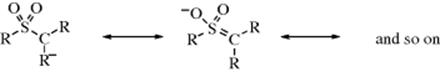
However, there is evidence against d-orbital overlap; and the stabilizing effects have been attributed to other causes.128 In the case of a PhS substituent, carbanion stabilization is thought to be due to a combination of the inductive and polarizability effects of the group, and d–pπ resonance and negative hyperconjugation play a minor role, if any.129 An α silicon atom also stabilizes carbanions.130
4. Field Effects. Most of the groups that stabilize carbanions by resonance effects (either the kind discussed in paragraph 1 above or the kind discussed in paragraph 3) have electron-withdrawing field effects and thereby stabilize the carbanion further by spreading the negative charge, although it is difficult to separate the field effect from the resonance effect. However, in a nitrogen (ylid R3N+–CR2, see Sec. 2.H), where a positive nitrogen is adjacent to the negatively charged carbon, only the field effect operates. Ylids are more stable than the corresponding simple carbanions. Carbanions are stabilized by a field effect if there is any heteroatom (O, N, or S) connected to the carbanionic carbon, provided that the heteroatom bears a positive charge in at least one important canonical form,131 for example,

5. Certain Carbanions Are Stable because They Are Aromatic. See the cyclopentadienyl anion in Section 2.I.ii, and other aromatic anions in Chapter 2.
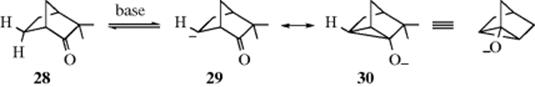
6. Stabilization by a Nonadjacent π Bond.132 In contrast to the situation with carbocations (see Sec. 2.C.i), there have been fewer reports of carbanions stabilized by interaction with a nonadjacent π bond. One that may be mentioned is 30, formed when optically active camphenilone (28) was treated with a strong base (potassium tert-butoxide).133 That 30 was truly formed was shown by the following facts: (1) A proton was abstracted: ordinary CH2 groups are not acidic enough for this base; (2) recovered 28 was racemized: 30 is symmetrical and can be attacked equally well from either side; (3) when the experiment was performed in deuterated solvent, the rate of deuterium uptake was equal to the rate of racemization; and (4) recovered 28 contained up to three atoms of deuterium per molecule, although if 29 were the only ion, no more than two could be taken up. Ions of this type, in which a negatively charged carbon is stabilized by a carbonyl group two carbons away, are called homoenolate ions.
Based on these four categories, functional groups in the α position stabilize carbanions in the following order: NO2 > RCO > COOR > SO2 > CN ~ CONH2 > halogen > H > R.
It is unlikely that free carbanions exist in solution, although some of the stabilized carbanions noted above have reasonable lifetimes in solution. Like carbocations, they usually exist as either ion pairs or they are solvated.134Among experiments that demonstrate ion pairing or solvation was the treatment of PhCOCHMe– M+ with ethyl iodide, where M+ was Li+, Na+, or K+. The half-lives of the reaction were135 for Li, 31 × 10−6; Na, 0.39 × 10−6; and K, 0.0045 × 10−6, demonstrating that the species involved were not identical. Similar results136 were obtained with Li, Na, and Cs triphenylmethides (Ph3C− M+).137 Where ion pairs are unimportant, carbanions are solvated. Cram105 demonstrated solvation of carbanions in many solvents. There may be a difference in the structure of a carbanion depending on whether it is free (e.g., in the gas phase) or in solution. The negative charge may be more localized in solution in order to maximize the electrostatic attraction to the counterion.138
The structure of simple unsubstituted carbanions is not known with certainty since they have not been isolated, but it is likely that the central carbon is sp3 hybridized, with the unshared pair occupying one apex of the tetrahedron. Carbanions are expected to have pyramidal structures (e.g., 31, similar to those of amines.
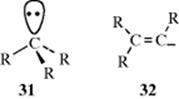
The methyl anion (CH3−) has been observed in the gas phase and reported to have a pyramidal structure.139 If this is taken as a general structure for carbanions, then any carbanion in which the three R groups are different should be chiral and reactions in which it is an intermediate should give retention of configuration. Attempts have been made to demonstrate this principle, but without success.140 A possible explanation is that pyramidal inversion takes place here, as in amines, so that the unshared pair and the central carbon rapidly oscillate from one side of the plane to the other. There is, however, other evidence for the sp3 nature of the central carbon and for its tetrahedral structure. Carbons at bridgeheads, although extremely reluctant to undergo reactions in which they must be converted to carbocations, easily undergo reactions in which they must be carbanions and stable bridgehead carbanions are known.141 Also, reactions at vinylic carbons proceed with retention,142 indicating that the intermediate 32 has sp2 hybridization and not the sp hybridization that would be expected in the analogous carbocation. A cyclopropyl anion can also hold its configuration.143
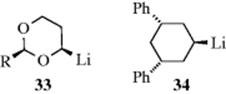
Carbanions in which the negative charge is stabilized by resonance involving overlap of the unshared-pair orbital with the π electrons of a multiple bond are essentially planar, as would be expected by the necessity for planarity in resonance, although unsymmetrical solvation or ion-pairing effects may cause the structure to deviate somewhat from true planarity.144 Cram144 showed that where chiral carbanions possessing this type of resonance are generated, retention, inversion, or racemization can result, depending on the solvent (see Sec. 12.A.ii). This result is explained by unsymmetrical solvation of planar or near-planar carbanions. However, some carbanions that are stabilized by adjacent sulfur or phosphorus, for example, are inherently chiral, since retention of configuration is observed where
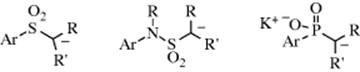
they are generated, even in solvents that cause racemization or inversion with other carbanions.145 It is known that in THF, PhCH(Li)Me behaves as a prochiral entity,146 and 33 has been prepared as an optically pure α-alkoxylithium reagent.147 Cyclohexyllithium (34) shows some configurational stability, and it is known that isomerization is slowed by an increase in the strength of lithium coordination and by an increase in solvent polarity.148It is known that a vinyl anion is configurationally stable whereas a vinyl radical is not. This is due to the instability of the radical anion that must be an intermediate for conversion of one isomer of vinyllithium to the other.149The configuration about the carbanionic carbon, at least for some of the α-sulfonyl carbanions, seems to be planar,150 and the inherent chirality is caused by lack of rotation about the C–S bond.151
5.B.ii. The Structure of Organometallic Compounds152
Whether a carbon–metal bond is ionic or polar-covalent is determined chiefly by the electronegativity of the metal and the structure of the organic part of the molecule. Ionic bonds become more likely as the negative charge on the metal-bearing carbon is decreased by resonance or field effects. Thus the sodium salt of acetoacetic ester has a more ionic carbon–sodium bond than methylsodium.
Most organometallic bonds are polar-covalent. Only the alkali metals have electronegativities low enough to form ionic bonds with carbon, and even here the behavior of lithium alkyls shows considerable covalent character. The simple alkyls and aryls of Na, K, Rb, and Cs153 are nonvolatile solids154 insoluble in benzene or other organic solvents, while alkyllithium reagents are soluble, although they too are generally nonvolatile solids. Organolithium reagents with alkyl units (alkyllithium reagents) do not exist as monomeric species in hydrocarbon solvents or ether.155 In benzene and cyclohexane, freezing-point-depression studies have shown that alkyllithium reagents are normally hexameric unless steric interactions favor tetrameric aggregates.156 Nuclear magnetic resonance studies, especially measurements of 13C–6Li coupling, have also shown aggregation in hydrocarbon solvents.157 Boiling-point-elevation studies have been performed in ether solutions, where alkyllithium reagents exist in two- to fivefold aggregates.158 Even in the gas phase159 and in the solid state,160 alkyllithium reagents exist as aggregates. X-ray crystallography has shown that methyllithium has the same tetrahedral structure in the solid state as in ether solution.160 However, tert-butyllithium is monomeric in THF, although dimeric in ether and tetrameric in hydrocarbon solvents.161 Neopentyllithium exists as a mixture of monomers and dimers in THF.162
The C–Mg bond in Grignard reagents is covalent and not ionic. The actual structure of Grignard reagents in solution has been a matter of much controversy over the years.163 In 1929, it was discovered164 that the addition of dioxane to an ethereal Grignard solution precipitates all the magnesium halide and leaves a solution of R2Mg in ether; (i.e., there can be no RMgX in the solution since there is no halide). The following equilibrium, now called the Schlenk equilibrium, was proposed as the composition of the Grignard solution:
![]()
in which 35 is a complex. Much work has demonstrated that the Schlenk equilibrium actually exists and that the position of the equilibrium depends on the identity of R, X, the solvent, the concentration, and the temperature.165 It has been known for many years that the magnesium in a solution of a Grignard reagent, no matter whether it is RMgX, R2Mg, or MgX2, can coordinate with two molecules of ether in addition to the two covalent bonds to generate the solvent-coordinated species shown.
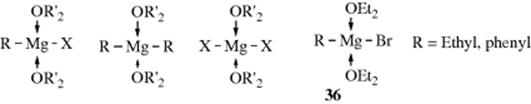
Rundle and Guggenberger166 performed X-ray diffraction studies on solid phenylmagnesium bromide dietherate and on ethylmagnesium bromide dietherate, which they obtained by cooling ordinary ethereal Grignard solutions until the solids crystallized. They found that the structures were magnesium bromides (e.g., 36). These solids still contained ether. When ordinary ethereal Grignard solutions167 prepared from bromomethane, chloromethane, bromoethane, and chloroethane were evaporated at ~100 °C under vacuum so that the solid remaining contained no ether, X-ray diffraction showed no RMgX, but a mixture of R2Mg and MgX2.168 These results indicate that in the presence of ether, RMgX√2Et2O is the preferred structure, while the loss of ether drives the Schlenk equilibrium to R2Mg + MgX2. However, conclusions drawn from a study of the solid materials do not necessarily apply to the structures in solution.
![]()
Boiling-point-elevation and freezing-point-depression measurements have demonstrated that in THF at all concentrations and in ether at low concentrations (up to ~0.1 M) Grignard reagents prepared from alkyl bromides and iodides are monomeric, (i.e., there are few or no molecules with two Mg atoms).169 Thus, part of the Schlenk equilibrium is operating but not the other part (i.e., 35 is not present in measurable amounts). This was substantiated by ![]() NMR spectra of the ethyl Grignard reagent in THF, which showed the presence of three peaks, corresponding to EtMgBr, Et2Mg, and MgBr2.170 That the equilibrium between RMgX and R2Mg lies far to the left for “ethylmagnesium bromide” in ether was shown by Smith and Becker,171 who mixed 0.1 M ethereal solutions of Et2Mg and MgBr2 and found that a reaction occurred with a heat evolution of 3.6 kcal mol−1 (15 kJ mol−1) of Et2Mg, and that the product was monomeric (by boiling-point elevation measurements). When either solution was added little by little to the other, there was a linear output of heat until almost a 1 : 1 molar ratio was reached. Addition of an excess of either reagent gave no further heat output. These results show that at least under some conditions the Grignard reagent is largely RMgX (coordinated with solvent), but that the equilibrium can be driven to R2Mg by evaporation of all the ether or by addition of dioxane.
NMR spectra of the ethyl Grignard reagent in THF, which showed the presence of three peaks, corresponding to EtMgBr, Et2Mg, and MgBr2.170 That the equilibrium between RMgX and R2Mg lies far to the left for “ethylmagnesium bromide” in ether was shown by Smith and Becker,171 who mixed 0.1 M ethereal solutions of Et2Mg and MgBr2 and found that a reaction occurred with a heat evolution of 3.6 kcal mol−1 (15 kJ mol−1) of Et2Mg, and that the product was monomeric (by boiling-point elevation measurements). When either solution was added little by little to the other, there was a linear output of heat until almost a 1 : 1 molar ratio was reached. Addition of an excess of either reagent gave no further heat output. These results show that at least under some conditions the Grignard reagent is largely RMgX (coordinated with solvent), but that the equilibrium can be driven to R2Mg by evaporation of all the ether or by addition of dioxane.
For some aryl Grignard reagents, it is possible to distinguish separate NMR chemical shifts for ArMgX and Ar2Mg.172 From the area under the peaks, it is possible to calculate the concentrations of the two species, and from them, equilibrium constants for the Schlenk equilibrium. These data show172 that the position of the equilibrium depends very markedly on the aryl group and the solvent, but that conventional aryl Grignard reagents in ether are largely ArMgX. In THF the predominance of ArMgX is less, and with some aryl groups there is actually more Ar2Mg present. Separate NMR chemical shifts have also been found for alkyl RMgBr and R2Mg in HMPA173 and in ether at low temperatures.174 When Grignard reagents from alkyl bromides or chlorides are prepared in triethylamine the predominant species is RMgX.175 Thus the most important factor determining the position of the Schlenk equilibrium is the solvent. For primary alkyl groups the equilibrium constant for the reaction as written above is lowest in Et3N, higher in ether, and still higher in THF.176
However, Grignard reagents prepared from alkyl bromides or iodides in ether at higher concentrations (0.5–1 M) contain dimers, trimers, and higher polymers, and those prepared from alkyl chlorides in ether at all concentrations are dimeric,177 so that 35 is in solution, probably in equilibrium with RMgX and R2Mg (i.e., the complete Schlenk equilibrium seems to be present).
The Grignard reagent prepared from 1-chloro-3,3-dimethylpentane in ether undergoes rapid inversion of configuration at the Mg containing carbon (demonstrated by NMR; this compound is not chiral).178 The mechanism of this inversion is not completely known. Despite the mechanistic ambiguity, in almost all cases, it is not possible to retain the configuration of a stereogenic carbon while forming a Grignard reagent.
Organolithium reagents (RLi) are very important reagents in organic chemistry. In recent years, a great deal has been learned about their structure179 in both the solid state and in solution. X-ray analysis of complexes of n-butyllithium with tetramethylethylenediamine (TMEDA), THF, and 1,2-dimethoxyethane (DME) shows them to be dimers and tetramers [e.g., (BuLi·DME)4];180 they are aggregates.181 X-ray analysis of isopropyllithium shows it to be a hexamer [(iPrLi)6],182 and unsolvated lithium aryls are tetramers.183 α-Ethoxyvinyllithium [CH2=C(OEt)Li] shows a polymeric structure with tetrameric subunits.184 Aminomethyl aryllithium reagents have been shown to be chelated and dimeric in solvents (e.g., THF).185 There are several functionalized organolithium reagents.186
The dimeric, tetrameric, and hexameric structures of organolithium reagents187 in the solid state is often retained in solution, but this is dependent on the solvent and complexing additives, if any. A tetrahedral organolithium compound is known,188 and the X-ray of an α,α-dilithio hydrocarbon has been reported.189 Phenyllithium is a mixture of tetramers and dimers in diethyl ether, but stoichiometric addition of THF, DME, or TMEDA leads to the dimer.190 The solution structures of mixed aggregates of butyllithium and amino-alkaloids has been determined191 as well as the solution structure of sulfur-stabilized allyllithium compounds.192 Vinyllithium is an 8:1 mixture of tetramer/dimer in THF at −90 °C, but addition of TMEDA changes the ratio of tetramer/dimer to 1 : 13 at −80 °C.193 Internally solvated allylic lithium compounds have been studied, showing the coordinated lithium to be closer to one of the terminal allyl carbons.194 A relative scale of organolithium stability has been established,195 and the issue of configurational stability of enantioenriched organolithium reagents has been examined.196
Enolate anions are an important class of carbanions that appear in a variety of important reactions, including alkylation α to a carbonyl group and the aldol (16-34) and Claisen condensation (16-85) reactions. Metal enolate anions of aldehydes, ketones, esters, and other acid derivatives exist as aggregates in ether solvents.197 There is evidence that the lithium enolate of isobutyrophenone is a tetramer in THF,198 but a dimer in DME.199 X-ray crystallography of ketone enolate anions have shown that they can exist as tetramers and hexamers.200 There is also evidence that the aggregate structure is preserved in solution and is probably the actual reactive species. Lithium enolate anions derived from esters are as dimers in the solid state201 that contain four THF molecules. It has also been established that the reactivity of enolate anions in alkylation and condensation reactions is influenced by the aggregate state of the enolate. It is also true that the relative proportions of (E) and (Z) enolate anions are influenced by the extent of solvation and the aggregation state. Addition of LiBr to a lithium enolate anion in THF suppresses the concentration of monomeric enolate.202Ab initio studies confirm the aggregate state of acetaldehyde.203 It is also known that α-Li benzonitrile [PhCH(Li)CN] exists as a dimer in ether and with TMEDA.204 Mixed aggregates of tert-butyllithium and lithium tert-butoxide are known to be hexameric.205
It might be mentioned that matters are much simpler for organometallic compounds with less-polar bonds. Thus Et2Hg and EtHgCl are both definite compounds, the former is a liquid and the latter is a solid. Organocalcium reagents are also known, and are formed from alkyl halides via a single electron transfer (SET) mechanism with free radical intermediates.206
5.B.iii. The Generation and Fate of Carbanions
There are two principal ways in which most carbanions are generated.
1. A Group Attached to a Carbon Leaves Without Its Electron Pair
![]()
The “leaving group” is most often a proton. In fact, the proton is removed by a suitable base, and this is a simple acid–base reaction.207 However, other leaving groups are known (see Chap 12), such as carboxyl:
![]()
2. A Negative Ion Adds to a Carbon–Carbon Double or Triple Bond (see Chapter 15)
![]()
The addition of a negative ion to a carbon–oxygen double bond (C=O) does not give a carbanion, but an alkoxide (R–O−), since the negative charge resides on the oxygen.
The most common reaction of carbanions is to donate electrons to a positive species, often a proton, or with another species that has an empty orbital in its outer shell (a Lewis acid–base reaction):
![]()
This means that carbanions react with electrophilic atoms (those functionalized so there is a δ+ carbon atom); see Chapter 16.
Carbanions may also form a bond with a carbon that already has four bonds, by displacing one of the four groups (SN2 reaction, see Chapter 10):
![]()
Like carbocations, carbanions can also react in ways in which they are converted to species that are still charged. They can add to double bonds (usually C=O double bonds; see Chapters 10 and 16),

or rearrange, although this is rare (see Chapter 18),
![]()
or they are oxidized to free radicals.208 A system in which a carbocation [Ph(p-Me2NC6H4)2C+] oxidizes a carbanion [(p-NO2C6H4)3C−] to give two free radicals, reversibly, so that all four species are present in equilibrium, has been demonstrated.209,210
Organometallic compounds that are not ionic but polar–covalent behave very much as if they were ionic and give similar reactions.