March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part I. Introduction
Chapter 7. Irradiation Processes in Organic Chemistry
Most reactions carried out in organic chemistry laboratories take place between molecules, all of which are in their ground electronic states. In a photochemical reaction,1 however, a reacting molecule has been previously promoted to an electronically excited state by absorption of light. A molecule in an excited state must lose its extra energy in some manner; it cannot remain in the excited state for long. The subject of electronic spectra is closely related to photochemistry. A chemical reaction is not the only possible means of relinquishing the extra energy in a photochemical process. In this chapter, electronically excited states and the processes of promotion to these states will be discussed. Reactions of such molecules are called photoreactions. There are enantioselective organocatalytic photoreactions, but they will not be discussed here.2 Two other methods are available to facilitate chemical reactions: sonochemistry and microwave chemistry. Although the physical processes involved are not the same excitation processes observed in photochemistry, irradiation with ultrasound or with microwaves have a significant influence on chemical reactivity, and both are widely used. For that reason, they are included in this chapter.
7.A. Photochemistry3
7.A.i. Excited States and the Ground State
Electrons can move from the ground-state energy level of a molecule to a higher level (i.e., an unoccupied orbital of higher energy) if outside energy is supplied. In a photochemical process, this energy is in the form of light. Light of any wavelength has an energy value associated with it given by E = hν, where n is the frequency of the light (n = velocity of light c divided by the wavelength λ), and h is Planck's constant. Since the energy levels of a molecule are quantized, the amount of energy required to raise an electron in a given molecule from one level to a higher one is a fixed quantity. Only light with exactly the frequency corresponding to this amount of energy will cause the electron to move to the higher level. If light of another frequency (too high or too low) is sent through a sample, it will pass out without a loss in intensity, since the molecules will not absorb it. However, if light of the correct frequency is passed into a sample, molecules will use that energy for electron promotion, and the light that leaves the sample will be diminished in intensity or altogether gone. A spectrophotometer is an instrument that allows light of a given frequency to pass through a sample and that detects (by means of a phototube) the amount of light that has been transmitted, that is, not absorbed. A spectrophotometer compares the intensity of the transmitted light with that of the incident light. Automatic instruments gradually and continuously change the frequency, and an automatic recorder plots a graph of absorption versus frequency or wavelength.
The energy of electronic transitions corresponds to light in the vis, UV, and far-UV regions of the spectrum (Fig. 7.1). Absorption positions are normally expressed in wavelength units, usually nanometers (nm).4 If a compound absorbs in the visible, it is colored, possessing a color complementary to that absorbed.5 Thus a compound absorbing in the violet has a yellow color. Organic chemists study the far-UV region less often than the vis or ordinary UV regions because special vacuum instruments are required, owing to the fact that oxygen and nitrogen absorb in these regions.
Fig. 7.1 The UV, vis, and IR portions of the electromagnetic spectrum.

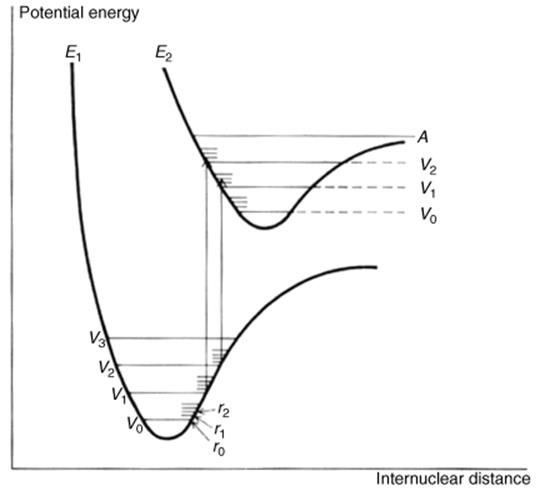
From these considerations it would seem that an electronic spectrum should consist of one or more sharp peaks, each corresponding to the transfer of an electron from one electronic level to another. Under ordinary conditions the peaks are seldom sharp. In order to understand why, it is necessary to realize that molecules are constantly vibrating and rotating and that these motions are also quantized. A molecule at any time is not only in a given electronic state, but also in a given vibrational and rotational state. The difference between two adjacent vibrational levels is much smaller than the difference between adjacent electronic levels, and the difference between adjacent rotational levels is smaller still. A typical situation is shown in Fig. 7.2. When an electron moves from one electronic level to another, it moves from a given vibrational and rotational level within that electronic level to some vibrational and rotational level at the next electronic level. A given sample contains a large number of molecules, and even if all of them are in the ground electronic state, they are still distributed among the vibrational and rotational states (though the ground vibrational state, Vo, is most heavily populated). This means that not just one wavelength of light will be absorbed, but a number of them close together, with the most probable transition causing the most intense peak. But in molecules containing more than a few atoms, there are so many possible transitions, and these are so close together that what is observed is a relatively broad band. The height of the peak depends on the number of molecules making the transition and is proportional to log ε, where ε is the extinction coefficient. The extinction coefficient can be expressed by ε = E/cl, where c is the concentration in moles per liter, l is the cell length in centimeters, and E = log Io/I, where Io is the intensity of the incident light and I of the transmitted light. The wavelength is usually reported as λmax, meaning that this is the top of the peak. Purely vibrational transitions (e.g., between Vo and V1 of E1), which require much less energy, are found in the IR region and are the basis of IR spectra. Purely rotational transitions are found in the far-IR and microwave (beyond the far-IR) regions.
Fig. 7.2 Energy curves for a diatomic molecule. Two possible transitions are shown. When an electron has been excited to the point marked A, the molecule may cleave (Sec. 7.A.v).
A UV or vis absorption peak is caused by the promotion of an electron in one orbital (usually ground-state) to a higher orbital. Normally, the amount of energy necessary to make this transition depends mostly on the nature of the two orbitals involved and much less on the rest of the molecule. Therefore, a simple functional group (e.g., the C=C double bond) always causes absorption in the same general area. A group that causes absorption is called a chromophore.
7.A.ii. Singlet and Triplet States: “Forbidden” Transitions
In most organic molecules, all electrons in the ground state are paired, with each member of a pair possessing opposite spin, as demanded by the Pauli principle. When one of a pair of electrons is promoted to an orbital of higher energy, the two electrons no longer share an orbital, and the promoted electron may, in principle, have the same spin as its former partner or the opposite spin. As seen in Chapter 5, a molecule in which two unpaired electrons have the same spin is called a triplet,6 while one in which all spins are paired is a singlet. Thus, at least in principle, for every excited singlet state there is a corresponding triplet state. In most cases, the triplet state has a lower energy than the corresponding singlet, which is in accord with Hund's rule. Therefore, a different amount of energy, and hence a different wavelength, is required to promote an electron from the ground state (which is almost always a singlet) to an excited singlet than to the corresponding triplet state.
It would thus seem that promotion of a given electron in a molecule could result either in a singlet or a triplet excited state depending on the amount of energy added. However, this is often not the case because transitions between energy levels are governed by selection rules, which state that certain transitions are “forbidden”. There are several types of “forbidden” transitions, two of which are more important than the others.
1. Spin-Forbidden Transitions. If the spin of an electron changes, transitions are not allowed, because a change from one spin to the opposite involves a change in angular momentum. Such a change would violate the law of conservation of angular momentum. Therefore, singlet–triplet and triplet–singlet transitions are forbidden, whereas singlet–singlet and triplet–triplet transitions are allowed.
2. Symmetry-Forbidden Transitions. Among the transitions in this class are those in which a molecule has a center of symmetry. In such cases, a g → g or u → u transition (see Sec. 1.A.) is “forbidden”, while a g → u or u → gtransition is allowed.
The word “forbidden” is in quotation marks because these transitions are not actually forbidden, but only highly improbable. In most cases, promotions from a singlet ground state to a triplet excited state are so improbable that they cannot be observed, and it is safe to state that in most molecules only singlet–singlet promotions take place. However, this rule does break down in certain cases, most often when a heavy atom (e.g., iodine) is present in the molecule, in which cases it can be shown from spectra that singlet–triplet promotions are occurring.7 Symmetry-forbidden transitions can frequently be observed, though usually with low intensity.
7.A.iii. Types of Excitation
When an electron in a molecule is promoted (normally only one electron in any molecule), it usually goes into the lowest available vacant orbital, though promotion to higher orbitals is also possible. For most organic molecules, there are consequently four types of electronic excitation:
1. σ → σ∗. Alkanes, which have no n or π electrons, can be excited only in this way.8
2. n → σ∗. Alcohols, amines,9 ethers, and so on can also be excited in this manner.
3. π → π∗. This pathway is open to alkenes as well as to aldehydes, carboxylic esters, and so on.
4. n → π∗. Aldehydes, ketones, carboxylic esters, and so on can undergo this promotion, as well as the other three.
The four excitation types above are listed in what is normally the order of decreasing energy. Thus light of the highest energy (in the far-UV) is necessary for σ → σ∗ excitation, while n → π∗ promotions are caused by ordinary UV light. However, the order may sometimes be altered in some solvents.
In 1,3-butadiene (and other compounds with two conjugated double bonds), there are two π and two π∗ orbitals (Sec. 2.C). The energy difference between the higher π (χ2) and the lower π∗ (χ3) orbital is less than the difference between the π and π∗ orbitals of ethylene. Therefore 1,3-butadiene requires less energy than ethylene, and thus light of a higher wavelength, to promote an electron. This is a general phenomenon, and it may be stated that, in general, the more conjugation in a molecule, the more the absorption is displaced toward higher wavelengths (see Table 7.1).10 When a chromophore absorbs at a certain wavelength and the substitution of one group for another causes absorption at a longer wavelength, a bathochromic shift is said to have occurred. The opposite kind of shift is called hypsochromic.
Table 7.1 Ultraviolet Absorption10 of CH3–(CH=CH)n–CH3 for Some Values of n
n
nm
2
227
3
263
6
352
9
413
Of the four excitation types listed above, the π → π∗ and n → π∗ are far more important in organic photochemistry than the other two. Compounds containing C=O groups can be excited in both ways, giving rise to at least two peaks in the UV.
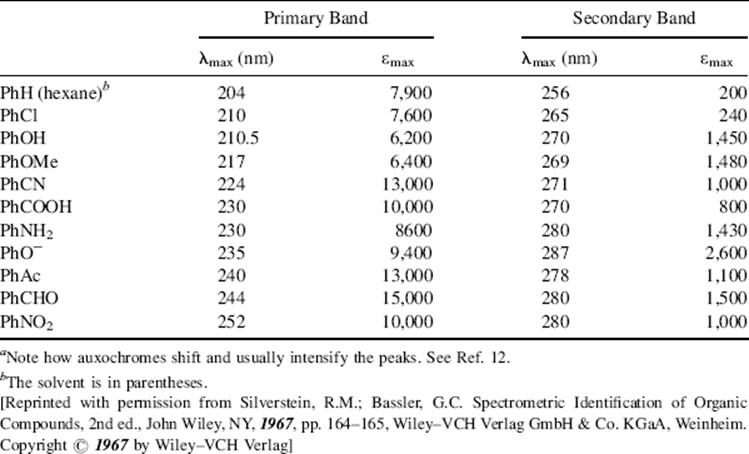
As seen above, a chromophore is a group that causes a molecule to absorb light. Examples of chromophores in the vis or UV are C=O, N=N,11 Ph, and NO2. Some chromophores in the far-UV (beyond 200 nm) are C=C, C≡C, Cl, and OH. An auxochrome is a group that displaces (through resonance) and usually intensifies the absorption of a chromophore present in the same molecule. Groups (e.g., Cl, OH, and NH2) are generally regarded as auxochromes since they shift (usually bathochromically) the UV and vis bands of chromophores (e.g., Ph or C=O; see Table 7.2).12 Since auxochromes are themselves chromophores (to be sure, generally in the far-UV), it is sometimes difficult to decide which group in a molecule is an auxochrome and which is a chromophore. For example, in acetophenone (PhCOMe) is the chromophore Ph or C=O? In such cases, the distinction becomes practically meaningless.
Table 7.2 Some UV Peaks of Substituted Benzenesa
7.A.iv. Nomenclature and Properties of Excited States
An excited state of a molecule can be regarded as a distinct chemical species, different from the ground state of the same molecule and from other excited states. It is obvious that some method of naming excited states is required. Unfortunately, there are several methods in use, depending on whether one is primarily interested in photochemistry, spectroscopy, or MO theory.13 One of the most common methods simply designates the original and newly occupied orbitals, with or without a superscript to indicate singlet or triplet. Thus the singlet state arising from promotion of a π to a π∗ orbital in ethylene would be the ![]() state or the π,π∗ singlet state. Another very common method can be used even in cases where one is not certain which orbitals are involved. The lowest-energy excited state is called S1, the next is S2, and so on. Triplet states are similarly labeled T1, T2, T3, and so on. In this notation, the ground state is So. Other notational systems exist, but this text shall discuss only the two types just mentioned.
state or the π,π∗ singlet state. Another very common method can be used even in cases where one is not certain which orbitals are involved. The lowest-energy excited state is called S1, the next is S2, and so on. Triplet states are similarly labeled T1, T2, T3, and so on. In this notation, the ground state is So. Other notational systems exist, but this text shall discuss only the two types just mentioned.
The properties of excited states are not easy to measure because of their generally short lifetimes and low concentrations, but enough work has been done for us to know that they often differ from the ground state in geometry, dipole moment, and acid or base strength.14 For example, acetylene, which is linear in the ground state, has a trans geometry in the excited state with ~sp2 carbons in the 1(π,π∗) state.15 Similarly, the ![]() and the
and the ![]() states of ethylene have a perpendicular and not a planar geometry,16 and the
states of ethylene have a perpendicular and not a planar geometry,16 and the ![]() and
and ![]() states of formaldehyde are both pyramidal.17 Triplet species tend to stabilize themselves by distortion, which relieves interaction between the unpaired electrons. Obviously, if the geometry is different, the dipole moment will probably differ also and the change in geometry and electron distribution often results in a change in acid or base strength.18 For example, the S1state of 2-naphthol is a much stronger acid (pK = 3.1) than the ground state (S0) of the same molecule (pK = 9.5).19
states of formaldehyde are both pyramidal.17 Triplet species tend to stabilize themselves by distortion, which relieves interaction between the unpaired electrons. Obviously, if the geometry is different, the dipole moment will probably differ also and the change in geometry and electron distribution often results in a change in acid or base strength.18 For example, the S1state of 2-naphthol is a much stronger acid (pK = 3.1) than the ground state (S0) of the same molecule (pK = 9.5).19
7.A.v. Photolytic Cleavage
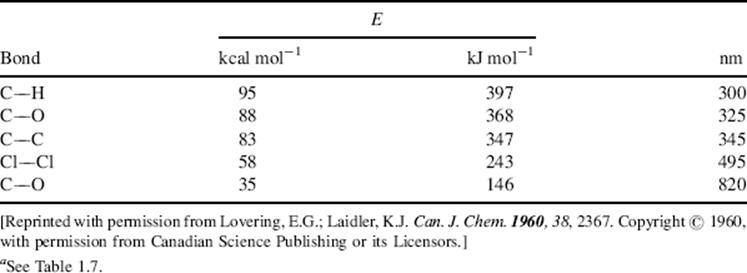
As stated above, when a molecule absorbs a quantum of light it is promoted to an excited state. Actually, that is not the only possible outcome. Because the energy of vis and UV light is of the same order of magnitude as that of covalent bonds (Table 7.3), another possibility is that the molecule may cleave into two parts, a process known as photolysis. There are three situations that can lead to cleavage:
Table 7.3 Typical Energies for Some Covalent Single Bondsa and the Corresponding Approximate Wavelengths.
1. The promotion may bring the molecule to a vibrational level so high that it lies above the right-hand portion of the E2 curve (line A in Fig. 7.2). In such a case, the excited molecule cleaves at its first vibration.
2. Even where the promotion is to a lower vibrational level, one that lies wholly within the E2 curve (e.g., V1 or V2), the molecule may still cleave. As shown in Fig. 7.2, equilibrium distances are greater in excited states than in the ground state. The Franck–Condon principle states that promotion of an electron takes place much faster than a single vibration (the promotion takes ~10−15 s; a vibration ~10−12 s). Therefore, when an electron is suddenly promoted, even to a low vibrational level, the distance between the atoms is essentially unchanged and the bond finds itself in a compressed condition like a pressed-in spring; this condition may be relieved by an outward surge that is sufficient to break the bond.
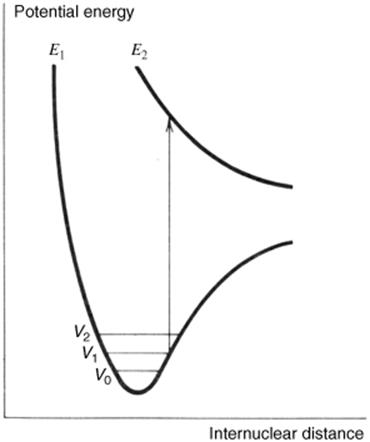
3. In some cases, the excited state is entirely dissociative (Fig. 7.3); that is, there is no distance where attraction outweighs repulsion, and the bond must cleave. An example is the hydrogen molecule, where a σ → σ∗ promotion always results in cleavage.
Fig. 7.3 Promotion to a dissociative state results in bond cleavage.
A photolytic cleavage can break the molecule into two smaller molecules or into two free radicals (see Sec. 7.A.vii). Cleavage into two ions, though known, is rare. Once free radicals are produced by a photolysis, they behave like free radicals produced in any other way (Chap 5) except that they may be in excited states, and this can cause differences in behavior.20
7.A.vi. The Fate of the Excited Molecule: Physical Processes
When a molecule has been photochemically promoted to an excited state, it does not remain in the excited state for long. Most promotions are from the S0 to the S1 state. As seen previously, promotions from So to triplet states are “forbidden”. Promotions to S2 and higher singlet states take place, but in liquids and solids these higher states usually drop very rapidly to the S1 state (~10−13 to ~10−11 s). The energy lost when an S2 or S3 molecule drops to S1 is given up in small increments to the environment by collisions with neighboring molecules. Such a process is called an energy cascade. In a similar manner, the initial excitation and the decay from higher singlet states initially populate many of the vibrational levels of S1, but these also cascade, down to the lowest vibrational level of S1. Therefore, in most cases, the lowest vibrational level of the S1 state is the only important excited singlet state.21 This state can undergo various physical and chemical processes. In the following list, we describe the physical pathways open to molecules in the S1 and excited triplet states. These pathways are also shown in a modified Jablonski diagram (Fig. 7.4) and in Table 7.4.
Fig. 7.4 Modified Jablonski diagram showing transitions between the excited and the ground state. Radiative processes are shown by straight lines, radiationless processes by wavy lines. vc = vibrational cascade; hνf = fluorescence; hνp = phosphorescence.
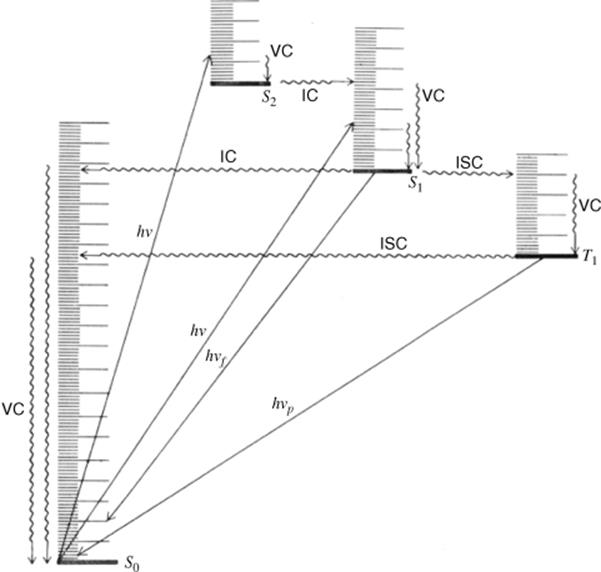
Table 7.4 Physical Processes Undergone by Excited Moleculesa
So + hν → ![]()
Excitation
![]()
![]()
S1 → S1 + hν
Fluorescence
S1
![]()
![]()
Intersystem crossing
![]()
Vibrational relaxation
T1 → So + hν
Phosphorescence
![]()
Intersystem crossing
![]()
Singlet–singlet transfer (photosensitization)
![]()
Triplet–triplet transfer (photosensitization)
a. The superscript v indicates vibrationally excited state: excited states higher than S1 or T1 are omitted.
1. A molecule in the S1 state can cascade down through the vibrational levels of the S0 state and thus return to the ground state by giving up its energy in small increments to the environment. This process is generally quite slow because the amount of energy is large and is called internal conversion (IC, see Fig. 7.4). Because it is slow, most molecules in the S1 state adopt other pathways.22
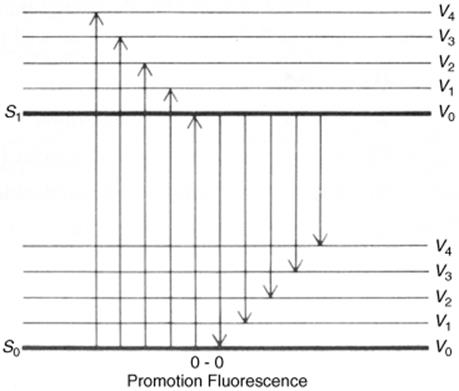
2. A molecule in the S1 state can drop to some low vibrational level of the So state all at once by giving off the energy in the form of light. This process, which generally happens within 10−9 s, is called fluorescence. This pathway is not very common either (because it is relatively slow), except for small molecules (e.g., diatomic) and rigid molecules (e.g., aromatic). For most other compounds, fluorescence is very weak or undetectable. For compounds that do fluoresce, the fluorescence emission spectra are usually the approximate mirror images of the absorption spectra. This comes about because the fluorescing molecules all drop from the lowest vibrational level of the S1 state to various vibrational levels of S0, while excitation is from the lowest vibrational level of So to various levels of S1 (Fig. 7.5). The only peak in common is the one that results from transitions between the lowest vibrational levels of the two states (called the 0–0 peak). In solution, even the 0–0 peak may be noncoincidental because the two states are solvated differently. Fluorescence nearly always arises from a S1 → So transition, although azulene (Sec. 2.I.iii) and its simple derivatives are exceptions,23 emitting fluorescence from S2 → So transitions.
Because of the possibility of fluorescence, any chemical reactions of the S1 state must take place very fast, or fluorescence will occur before they can happen.
3. Most molecules (but by no means all) in the S1 state can undergo an intersystem crossing (ISC, see Fig. 7.4) to the lowest triplet state T1.24 An important example is benzophenone, of which 100% of the molecules that are excited to the S1 state cross over to the T1.25 Intersystem crossing from singlet to triplet is of course a “forbidden” pathway, since the angular-momentum problem (Sec. 7.A.ii) must be taken care of, but this often takes place by compensations elsewhere in the system. Intersystem crossings take place without loss of energy. Since a singlet state usually has a higher energy than the corresponding triplet, this means that energy must be given up. One way for this to happen is for the S1 molecule to cross to a T1 state at a high vibrational level and then for the T1 to cascade down to its lowest vibrational level (see Fig. 7.4). This cascade is very rapid (10−12 s). When T2 or higher states are populated, they too rapidly cascade to the lowest vibrational level of the T1 state.
4. A molecule in the T1 state may return to the So state by giving up heat (ISC) or light (this is called phosphorescence).26 Of course, the angular momentum difficulty exists here, so that both ISC and phosphorescence are very slow (~10−3–101 s). This means that T1 states generally have much longer lifetimes than S1 states. When they occur in the same molecule, phosphorescence is found at lower frequencies than fluorescence (because of the higher difference in energy between S1 and So than between T1 and So) and is longer-lived (because of the longer lifetime of the T1 state).
5. If nothing else happens to it first, a molecule in an excited state (S1 or T1) may transfer its excess energy all at once to another molecule in the environment, in a process called photosensitization.27 The excited molecule, which we will call D for donor, thus drops to So while the other molecule (A for acceptor) becomes excited:
![]()
Thus there are two ways for a molecule to reach an excited state: by absorption of a quantum of light or by transfer from a previously excited molecule.28 The donor D is also called a photosensitizer. This energy transfer is subject to the Wigner spin-conservation rule, which is actually a special case of the law of conservation of momentum we encountered previously. According to the Wigner rule, the total electron spin does not change after the energy transfer. For example, when a triplet species interacts with a singlet these are some allowed possibilities:29
In all these cases, the products have three electrons spinning “up” and the fourth “down” (as do the starting molecules). However, formation of, say, two triplets (↑↓ + ↓↓) or two singlets (↑↓ + ↑↓), whether ground states or excited, would violate the rule.
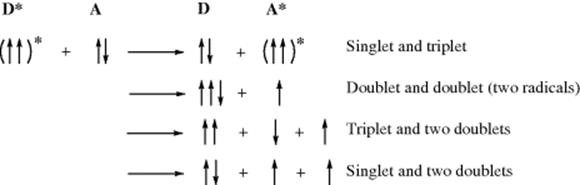
In the two most important types of photosensitization, both of which are in accord with the Wigner rule, a triplet excited state generates another triplet and a singlet generates a singlet:
![]()
Singlet–singlet transfer can take place over relatively long distances (e.g., 40 Å), but triplet transfer normally requires a collision between the molecules.30 Both types of photosensitization can be useful for creating excited states when they are difficult to achieve by direct irradiation. Photosensitization is therefore an important method for carrying out photochemical reactions when a molecule cannot be brought to the desired excited state by direct absorption of light. Triplet–triplet transfer is especially important because triplet states are usually much more difficult to prepare by direct irradiation than singlet states (often impossible) and because triplet states, having longer lifetimes, are much more likely than singlets to transfer energy by photosensitization. Photosensitization can also be accomplished by electron transfer.31
In choosing a photosensitizer,32 one should avoid a compound that absorbs in the same region as the acceptor because the latter will then compete for the light.33 For examples of the use of photosensitization to accomplish reactions, see Reactions 15-62 and 15-63.
6. An excited species can be quenched. Quenching is the deactivation of an excited molecular entity intermolecularly by an external environmental influence (e.g., a quencher), or intramolecularly by a substituent through a nonradiative process.34 When the external environmental influence (quencher) interferes with the behavior of the excited state after its formation, the process is referred to as dynamic quenching. Common mechanisms include energy transfer, charge transfer, and so on. When the environmental influence inhibits the excited state formation, the process is referred to as static quenching. A quencher is defined as a molecular entity that deactivates (quenches) an excited state of another molecular entity, either by energy transfer, electron transfer, or by a chemical mechanism.34
An example is the rapid triplet quenching of aromatic ketone triplets35 by amines, which is well known.36 Alkyl and aryl thiols and thioethers also serve as quenchers in this system.37 In this latter case, the mechanism involves electron transfer from the sulfur atom to the triplet ketone, and this is supported by theoretical calculations.38 Aromatic ketone triplets are quenched by phenols, and the photochemical reaction between aromatic ketones and phenols is efficient only in the presence of an acid catalyst.39 Indirect evidence has been provided for involvement of the hydrogen-bonded triplet exciplex and for the role of electron transfer in this reaction.40
Fig. 7.5 Promotion and fluorescence between S1 and S0 states.
7.A.vii. The Fate of the Excited Molecule: Chemical Processes
Although both excited singlet and triplet species can undergo chemical reactions, they are much more common for triplets, simply because these generally have much longer lifetimes. Excited singlet species, in most cases, have a lifetime of <10−10 s and undergo one of the physical processes already discussed before they have a chance to react chemically. Therefore, photochemistry is largely the chemistry of triplet states.41 Table 7.542 lists many of the possible chemical pathways that can be taken by an excited molecule.43 The first four of these are unimolecular reactions; the others are bimolecular. In the case of bimolecular reactions, it is rare for two excited molecules to react with each other (because the concentration of excited molecules at any one time is generally low); reactions are between an excited molecule and an unexcited molecule of either the same or another species. The reactions listed in Table 7.5 are primary processes. Secondary reactions often follow, since the primary products are frequently radicals or carbenes; even if they are ordinary molecules, they are often in upper vibrational levels and so have excess energy. In almost all cases, the primary products of photochemical reactions are in their ground states, though exceptions are known.44 Of the reactions listed in Table 7.5, the most common are cleavage into radicals (1), decomposition into molecules (2), and (in the presence of a suitable acceptor molecule) photosensitization (7), which we have already discussed. The following are some specific examples of reaction categories (1)–(6). Other examples are discussed in Part II.45,46
Table 7.5 Primary Photochemical Reactionsa of an Excited Molecule A–B–Cb
Reactions
Reaction Type
Example Number
(A–B–C) → A–B• + C•
Simple cleavage into radicals46
(1)
(A–B–C) → E + F
Decomposition into molecules
(2)
(A–B–C) → A–C–B
Intramolecular rearrangement
(3)
(A–B–C) → A–B–C′
Photoisomerization
(4)
(A–B–C) + RH → A–B–C–H + R•
Hydrogen-atom abstraction
(5)
(A–B–C) → (ABD)2
Photodimerization
(6)
(A–B–C) + A → ABX + A∗
Photosensitization
(7)
a. Examples are given in the text; the most common are (1), (2), and, in the presence of a suitable acceptor molecule (7).
b. See Ref. 42.
Category 1. Simple Cleavage into Radicals.47 Aldehydes and ketones absorb in the 230–330-nm region. This is assumed to result from an n → π∗ singlet–singlet transition. The excited aldehyde or ketone can then cleave.48
![]()
When applied to ketones, this is called Norrish Type I cleavage or often just Type I cleavage. In a secondary process, the acyl radical (R′–CO•) can then lose CO to give R′• radicals. Another example of a category 1 process is cleavage of Cl2 to give two Cl atoms. Other bonds that are easily cleaved by photolysis are the O–O bonds of peroxy compounds and the C–N bonds of aliphatic azo compounds (R–N=N–R).49 The latter is an important source of radicals R•, since the other product is the very stable N2.
Category 2. Decomposition into Molecules. Aldehydes (though not generally ketones) can also cleave in this manner:
![]()
This is an extrusion reaction (see Chapter 17). In another example of a process in category 2, aldehydes and ketones with an γ hydrogen can cleave in still another way (a β-elimination, see Chap 17):
![]()
This reaction, called Norrish Type II cleavage,50 involves intramolecular abstraction of the γ hydrogen followed by cleavage of the resulting diradical51 (a secondary reaction) to give an enol that tautomerizes to the aldehyde or ketone product.52
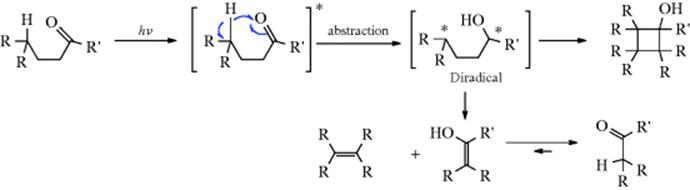
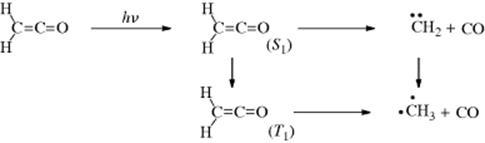
Both singlet and triplet n,π∗ states undergo the reaction.53 The intermediate diradical can also cyclize to a cyclobutanol, which is often a side product. Carboxylic esters, anhydrides, and other carbonyl compounds can also give this reaction.54 The photolysis of ketene to CH2 (Sec. 5.D.ii) is still another example of a reaction in category 2. Both singlet and triplet CH2 are generated, the latter in two ways:
Reactions are known where both Norrish Type I and Type II reactions compete, and the substituents on and nature of the substrate will determine which leads to the major product.55
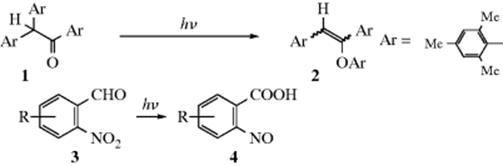
Category 3. Intramolecular Rearrangement. Two examples are the rearrangement of the trimesityl compound 1 to the enol ether (2),56 and irradiation of o-nitrobenzaldehydes (3) to give o-nitrosobenzoic acids (4).57
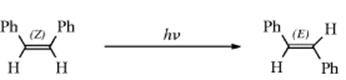
Category 4. Photoisomerization. The most common reaction in this category is photochemical cis–trans isomerization.58 For example, cis-stilbene can be converted to the trans isomer,59 and the photoisomerization of O-methyl oximes is known.60
The isomerization takes place because the excited states, both S1 and T1, of many alkenes have a perpendicular instead of a planar geometry (Sec. 7.A.iv), so cis–trans isomerism disappears upon excitation. When the excited molecule drops back to the So state, either isomer can be formed. A useful example is the photochemical conversion of cis-cyclooctene to the much less stable trans isomer.61 Another interesting example of this isomerization involves azo crown ethers. The crown ether 5, in which the N=N bond is anti, preferentially binds NH4+, Li+, and Na+, but the syn isomer preferentially binds K+ and Rb+ (see Sec. 3.C.ii). Thus, ions can be selectively put in or taken out of solution merely by turning a light source on or off.62
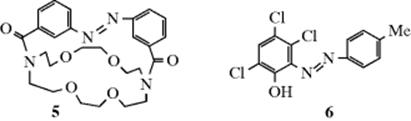
In another example, the trans azo compound (6) is converted to its cis isomer when exposed to light. In this case63 the cis isomer is a stronger acid than the trans. The trans isomer is dissolved in a system containing a base, wherein a liquid membrane separates two sides, one of which is illuminated, the other kept dark. On the illuminated side, the light converts the trans isomer to the cis. The cis isomer, being a stronger acid, donates its proton to the base, converting cis-ArOH to cis-ArO−. This ion migrates to the dark side, where it rapidly reverts to the trans ion, which reacquires a proton. Because each cycle forms one H3O+ ion in the illuminated compartment and one ![]() ion in the dark compartment, the process reverses the normal reaction whereby these ions neutralize each other.64 Thus the energy of light is used to do chemical work.65 Another example of a category 4 reaction is the conversion of bicyclo[2.2.1]hept-2,5-diene to 7.58 The thermal isomerization of dibenzosemibullvalene (9) to the corresponding dibenzodihydropentalenofuran (8) in quantitative yield was known,66 but in another example of a category 4 reaction the photochemical isomerization of 8 to 9 has now been reported.67
ion in the dark compartment, the process reverses the normal reaction whereby these ions neutralize each other.64 Thus the energy of light is used to do chemical work.65 Another example of a category 4 reaction is the conversion of bicyclo[2.2.1]hept-2,5-diene to 7.58 The thermal isomerization of dibenzosemibullvalene (9) to the corresponding dibenzodihydropentalenofuran (8) in quantitative yield was known,66 but in another example of a category 4 reaction the photochemical isomerization of 8 to 9 has now been reported.67
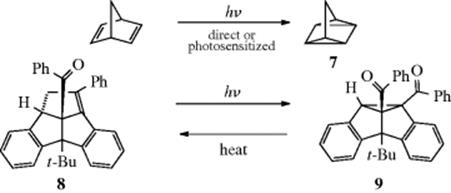
These examples illustrate that the use of photochemical reactions can make it very easy to obtain compounds that would be difficult to get in other ways. Reactions similar to these are discussed in Reaction 15-63.
Category 5. Hydrogen-Atom Abstraction. When benzophenone is irradiated in isopropyl alcohol, the initially formed S1 state crosses to the T1 state, which abstracts hydrogen from the solvent to give the radical 10. Radical 10then abstracts another hydrogen to give benzhydrol (11) or dimerizes to benzpinacol (12):
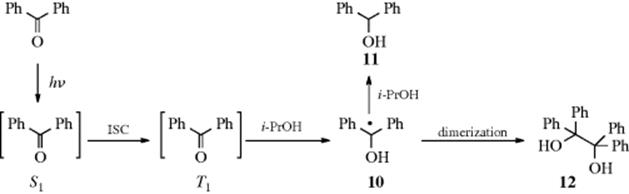
An example of intramolecular abstraction has already been given (see category 2 in this section).
Category 6. Photodimerization. An example is dimerization of cyclopentenone:68
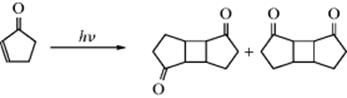
See Reaction 15-63 for a discussion of this and similar reactions.
7.A.viii. The Determination of Photochemical Mechanisms69
The methods used for the determination of photochemical mechanisms are largely the same as those used for organic mechanisms in general (Chapter 6): product identification, isotopic tracing, the detection and trapping of intermediates, and kinetics. There are, however, a few new factors: (1) there are generally many products in a photochemical reaction, as many as 10 or 15; (2) in measuring kinetics, there are more variables, since it is possible to study the effect on the rate of the intensity or the wavelength of light; (3) in the detection of intermediates by spectra the technique of flash photolysis can be used, which can detect extremely short-lived intermediates.
In addition to these methods, there are two additional techniques.
1. The use of emission (fluorescence and phosphorescence), as well as absorption spectroscopy. From these spectra the presence of as well as the energy and lifetime of singlet and triplet excited states can often be calculated.
2. The study of quantum yields. The quantum yield is the fraction of absorbed light that goes to produce a particular result. There are several types. A primary quantum yield for a particular process is the fraction of molecules absorbing light that undergo that particular process. Thus, if 10% of all the molecules that are excited to the S1 state cross over to the T1 state, the primary quantum yield for that process is 0.10. However, primary quantum yields are often difficult to measure. A product quantum yield (usually designated Φ) for a product P that is formed from a photoreaction of an initially excited molecule A can be expressed as:
![]()
Product quantum yields are much easier to measure. The number of quanta absorbed can be determined by an instrument called an actinometer, which is actually a standard photochemical system whose quantum yield is known. An example of the information that can be learned from quantum yields is the following. If the quantum yield of a product is finite and invariant with changes in experimental conditions, it is likely that the product is formed in a primary rate-determining process. Another example: In some reactions, the product quantum yields are found to be well over 1 (perhaps as high as 1000). Such a finding indicates a chain reaction (see Sec. 14.A.i for a discussion of chain reactions).