March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part I. Introduction
Chapter 8. Acids and Bases
Two acid–base theories are used in organic chemistry today: the Br![]() nsted theory and the Lewis theory.1 These theories are quite compatible and are used for different purposes.2 However, the Lewis-based idea of electron-donating species (bases) and electron- accepting species (acids) is often the more useful for organic chemistry. Remember also that most organic reactions are not done in an aqueous medium, and focus on electron transfer rather than proton transfer is far more useful.
nsted theory and the Lewis theory.1 These theories are quite compatible and are used for different purposes.2 However, the Lewis-based idea of electron-donating species (bases) and electron- accepting species (acids) is often the more useful for organic chemistry. Remember also that most organic reactions are not done in an aqueous medium, and focus on electron transfer rather than proton transfer is far more useful.
8.A. Brønsted Theory
According to this theory, an acid is defined as a proton donor3 and a base as a proton acceptor. However, a base must have a pair of electrons available to share with the proton; this is usually present as an unshared pair, but sometimes is in a π orbital. By this definition, an acid–base reaction is the transfer of a proton from an acid to a base. However, protons do not exist free in solution, but must be attached to an electron pair. In fact, the acid does not “give up” a proton, but rather the base donates electrons to the proton, “pulling it away” to form the conjugate acid. After removal of the proton, the species remaining (the conjugate base) still retains the electron pair to which the proton was formerly attached. The conjugate base, in theory at least, can reacquire a proton and is therefore a base. All acids will generate a conjugate base upon reaction with a suitable base, and all bases will generate a conjugate acid by reaction with a suitable acid. All acid–base reactions fit the equation
![]()
No charges are shown in this equation, but an acid always has a charge one positive unit higher than that of its conjugate base.
8.A.i Br![]() nsted Acids
nsted Acids
According to the Br![]() nsted definition, acid strength may be defined as the tendency to give up a proton and base strength as the tendency to accept a proton. All acid–base reactions are reversible, and both an acid and a conjugate acid are present in the equilibrium mixture. In one sense, acid–base reactions occur because the acid and the conjugate acid are not of equal strength (i.e., the equilibrium can be shifted to one side or the other). If an acid, say HCl, is placed in contact with the conjugate base of a weaker acid, say acetate ion, the conjugate acid in this reaction would be acetic acid. Since HCl is a stronger acid than acetic acid (see Table 8.1), the equilibrium lies well to the right. As the reaction is written, if the equilibrium lies to the right (higher concentration of acetic acid and a lower concentration of HCl), HCl is the stronger acid. Likewise, acetate is taken to be a stronger base than the chloride ion. If this is a correct statement, treatment of acetic acid with chloride ion should give essentially no reaction, since the weaker acid already has the proton. This is found to be correct.
nsted definition, acid strength may be defined as the tendency to give up a proton and base strength as the tendency to accept a proton. All acid–base reactions are reversible, and both an acid and a conjugate acid are present in the equilibrium mixture. In one sense, acid–base reactions occur because the acid and the conjugate acid are not of equal strength (i.e., the equilibrium can be shifted to one side or the other). If an acid, say HCl, is placed in contact with the conjugate base of a weaker acid, say acetate ion, the conjugate acid in this reaction would be acetic acid. Since HCl is a stronger acid than acetic acid (see Table 8.1), the equilibrium lies well to the right. As the reaction is written, if the equilibrium lies to the right (higher concentration of acetic acid and a lower concentration of HCl), HCl is the stronger acid. Likewise, acetate is taken to be a stronger base than the chloride ion. If this is a correct statement, treatment of acetic acid with chloride ion should give essentially no reaction, since the weaker acid already has the proton. This is found to be correct.
![]()
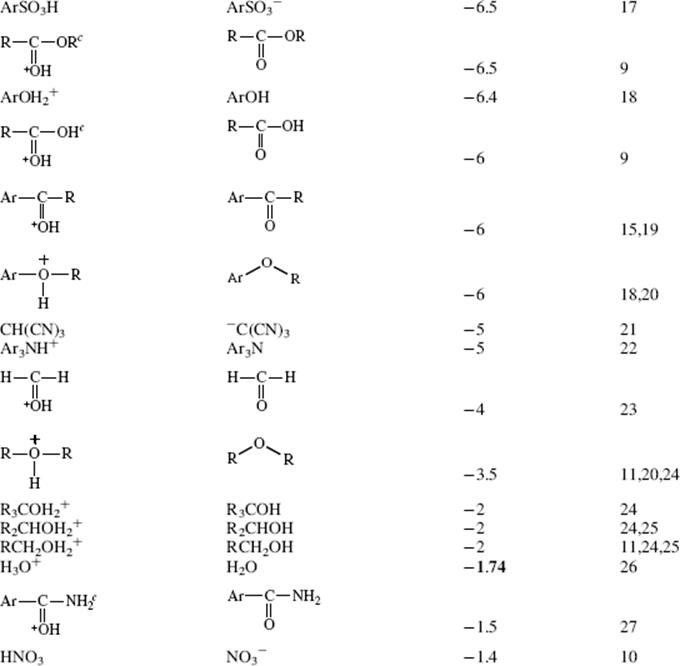
Table 8.1 The pKa Values for Many Types of Acidsa

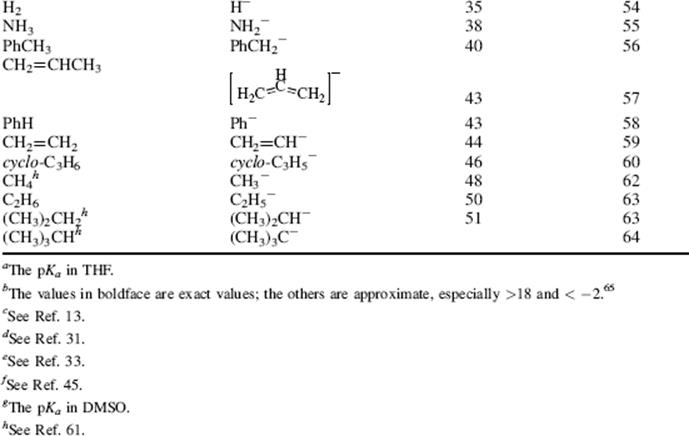
For a comparison of two different acids, the position of the equilibrium in reaction with a common base allows the relative strengths of acids to be determined.4 Likewise, the strength of two different bases will be determined by comparing the equilibrium established when they react with a common acid. By definition, the acid and base are always drawn on the left side of the equation, and the conjugate acid and conjugate base are assumed to be on the right side of the equation.
Of course, if the two acids involved are close to each other in strength, a measurable reaction will occur from both sides. This finding really means that the concentration of acid and base at equilibrium will be close to that of the concentration of the conjugate acid and conjugate base. However, the position of equilibrium will still be over to the side of the weaker acid (unless the acidities are equal within experimental limits). If the concentration of acid and base is higher, the reaction of conjugate acid and conjugate base is more facile, and the compound labeled as the acid is considered to be a weaker acid. If the concentration of the conjugate acid and conjugate base is higher, the reaction of the acid and base is more facile, and the compound labeled as the acid is a stronger acid.
Using these protocols as the definition of acid strength, it is possible to construct a table in which acids are listed in order of acid strength5 (Table 8.1).6 The conjugate base is shown next to each acid in Table 8.1. Using the axiom that a strong acid generates a weak conjugate base and a weak acid will generate a strong conjugate base, it is clear that if the acids in such a table are listed in decreasing order of acid strength, the bases must be listed in increasingorder of base strength. The pKa values7 in Table 8.1 are most accurate in the middle of the table.8–67
The pKa values are much harder to measure68 for very strong and very weak acids, and these values must be regarded as approximate. If one did not have the pKa values available, it can be determined experimentally that HClO4 is a stronger acid than H2SO4. A mixture of HClO4 and H2SO4 in 4-methyl-2-pentanone can be titrated to an HClO4 end point without interference by H2SO4.69 Similarly, HClO4 can be shown to be stronger than HNO3 or HCl. However, this is not quantitative, and the value of −10 in the table is not much more than an educated guess. The values for RNO2H+, ArNO2H+, HI, RCNH+ and RSH2+ must also be regarded as highly speculative.70 A wide variety of pKavalues have been reported for the conjugate acids of even such simple bases as acetone67 (−0.24 to −7.2), diethyl ether (−0.30 to −6.2), ethanol (−0.33 to −4.8), methanol (−0.34 to −4.9), and 2-propanol (−0.35 to −5.2), depending on the method used to measure them.71 Very accurate values can be obtained only for acids weaker than hydronium ion and stronger than water.
A crystallographic scale of acidity has been developed, including the acidity of C–H compounds. Measuring the mean C–H![]() O distances in crystal structures correlated well with conventional pKa(DMSO) values,72 where DMSO is dimethyl sulfoxide. An ab initio study was able to correlate ring strain in strained hydrocarbons with hydrogen-bond acidity.73 The kinetic acidity of aliphatic hydrocarbons has been determined.74
O distances in crystal structures correlated well with conventional pKa(DMSO) values,72 where DMSO is dimethyl sulfoxide. An ab initio study was able to correlate ring strain in strained hydrocarbons with hydrogen-bond acidity.73 The kinetic acidity of aliphatic hydrocarbons has been determined.74
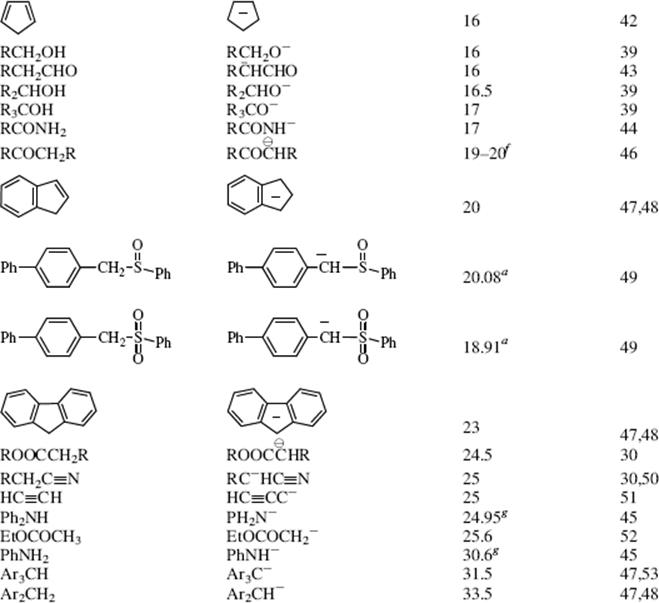
The bottom portion of Table 8.1 consists of very weak acids (pKa above that of water ≈15.8).75 In most of these acids, the proton is lost from a carbon atom, and such acids are known as carbon acids. The pKa values for such weak acids are often difficult to measure and are known only approximately. The methods used to determine the relative positions of these acids are discussed in Chapter 5.76 The acidity of carbon acids is proportional to the stability of the carbanions that are their conjugate bases (see Sec. 5.B.i).
The extremely strong acids at the top of the table are known as superacids (see Sec. 5.A.ii).77 The actual species present in the FSO3H–SbF5 mixture are probably H[SbF5(SO3F)] and H[SbF2(SO3F)4.66 The addition of SO3 causes formation of the still stronger H[SbF4(SO3F)2], H[SbF3(SO3F)3], and H[(SbF5)2(SO3F)].66 There is a study of electrophilic intermediates that are generated in superacids78 (also see Chapter 10).
By the use of tables (e.g., Table 8.1), it is possible to determine whether a given acid will react with a given base to give reasonable concentrations of the conjugate acid and base. For tables in which acids are listed in order of decreasing strength, the rule is that any acid will react with any base in the table that is below it but not with any above it.79 The greater the separation in the table, the better the reaction. It must be emphasized that the order of acid strength in Table 8.1 separation applies when a given acid and base react without a solvent or, when possible, in water. In other solvents, the order may be greatly different (see Sec. 8.G). In the gas phase, where solvation effects are completely or almost completely absent, acidity orders may also differ greatly.80 For example, in the gas phase, toluene is a stronger acid than water and tert-butoxide ion is a weaker base than methoxide ion81 (see also Sec. 8.G). It is also possible for the acidity order to change with temperature. For example, >50 °C the order of base strength is BuOH > H2O > Bu2O; from 1 to 50 °C the order is BuOH > Bu2O > H2O; while <1 °C the order becomes Bu2O > BuOH > H2O.82
8.A.ii. Br![]() nsted Bases
nsted Bases
Basicity may be measured by a parameter known as proton affinity of an anion. The dissociation of a hydrogen ion for a molecule in the gas phase is called the proton affinity of the conjugate base.83 A hydrogen-bond basicity scale has been developed that can be used to determine the relative basicity of molecules. Table 8.2 gives the pKHB values for several common heteroatom-containing molecules.84 This is obtained from the protonated form (conjugated acid) of the base in question. The larger the number, the more basic is that compound. The basicity of aliphatic amines has been calculated,85 the ion-pair basicity of amines in THF86 and in water87 has been determined, and the basicity of pyridine was examined.88 There are secondary deuterium isotope effects for measuring the basicity of secondary amines, and deuteration was found to increase the basicity.89 Weaker bases have also been examined, and the basicity of carbonyl compounds in carbon tetrachloride has been determined.97 Alkenes are weak bases98 that react with strong acids (e.g., HCl or HBr, Reaction 15-02). Note that extremely twisted amides (Sec. 4.Q.ii) exhibit high basicity.99
Table 8.2 The pKHB Values for Many Types of Bases.
Base
Approximate pKHB
Reference
N-Methyl-2-piperidone
2.60
90
Et2NCONEt2
2.43
90
N-Methyl-2-pyrrolidinone
2.38
90
PhCONMe2
2.23
90
HCONMe2
2.10
90
PhCONHMe
2.03
90
18-crown-6
1.98
91
HCONHMe
1.96
90
Aniline
4.60
92
N-methylaniline
4.85
92
PhNHNH2
5.27
92
Ph(Me)NNH2
4.99
92
15-crown-5
1.82
91
12-crown-4
1.73
91
PhOCONMe2
1.70
90
Et2N–CN
1.63
93
Me2N–CN
1.56
93
δ-Valerolactone
1.43
94
Oxetane
1.36
91
γ-Butyrolactone
1.32
94
THF
1.28
91
Cyclopentanone
1.27
95
t-BuOMe
1.19
91
Acetone
1.18
95
MeCOOEt
1.07
95
1,4-Dioxane
1.03
91
Et2O
1.01
91
1,3-Dioxane
0.93
91
1-Methyloxirane
0.97
91
PhCOOMe
0.89
94
MeOCOOMe
0.82
94
PhCHO
0.78
95
Bu2O
0.75
91
HCOOEt
0.66
94
MeCHO
0.65
95
Me2NO2
0.41
96
MeNO2
0.27
96
PhNO2
0.30
96
Furan
−0.40
91
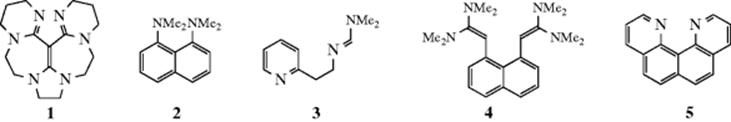
A class of organic compounds termed superbases has been developed.100 Vinamidine type or Schwesinger proton sponges (see Sec. 8.F), 1,101 are dubbed superbases and are probably the most powerful organic neutral bases known. The pKa (pKBH+) in MeCN was measure as 31.94. It has been shown that the pKa values of strong neutral organic (super)bases in acetonitrile are well described by the density functional theory.102 The fundamental type of proton sponge is 1,8-bis(dimethylamino)naphthalene (2, see Sec. 8.F), with a pKBH+ of 18.18.103 Other superbase-type compounds include amidinazines [e.g., N1,N1-dimethyl-N2-β-(2-pyridylethyl)-formamidine (3)], pKBH+ in DMSO = 25.1,104 1,8-bis(tetramethylguanidino)naphthalene, (4),105 and quinolino[7,8-h]quinolines (e.g., 5) with a pKBH+ = 12.8.106
It is important to note that organometallic compounds, such as Grignard reagents (RMgX) and organolithium reagents (RLi),107 are powerful bases. The conjugate bases of both of these bases are alkanes, (R–H), which are very weak acids indeed (see Table 8.1).