March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 11. Aromatic Substitution, Electrophilic
Most substitutions at an aliphatic carbon are by nucleophiles. In aromatic systems the situation is reversed, because the high electron density at the aromatic ring leads to its reactivity as a Lewis base or a Br![]() nsted–Lowry base, depending on the positive species. In electrophilic substitutions, a positive ion or the positive end of a dipole or induced dipole is attacked by the aromatic ring. The leaving group (the electrofuge) must necessarily depart without its electron pair. In nucleophilic substitution reactions, the chief leaving groups are those best able to carry the unshared pair: Br−, H2O, OTs−, and so on (i.e., the weakest bases). In electrophilic substitution reactions, the most important leaving groups are those that can best exist without the pair of electrons necessary to fill the outer shell; that is, the weakest Lewis acids. The influence of solvents will vary with the reaction in many cases, and such details are discussed where appropriate in the reactions section (Sec. 11.F).1
nsted–Lowry base, depending on the positive species. In electrophilic substitutions, a positive ion or the positive end of a dipole or induced dipole is attacked by the aromatic ring. The leaving group (the electrofuge) must necessarily depart without its electron pair. In nucleophilic substitution reactions, the chief leaving groups are those best able to carry the unshared pair: Br−, H2O, OTs−, and so on (i.e., the weakest bases). In electrophilic substitution reactions, the most important leaving groups are those that can best exist without the pair of electrons necessary to fill the outer shell; that is, the weakest Lewis acids. The influence of solvents will vary with the reaction in many cases, and such details are discussed where appropriate in the reactions section (Sec. 11.F).1
11.A. Mechanisms
Electrophilic aromatic substitutions are unlike nucleophilic substitutions in that the large majority proceeds by just one mechanism with respect to the substrate.2 In this mechanism, called the arenium ion mechanism, the electrophile, which can be viewed as a Lewis acid, is attacked by the π electrons of the aromatic ring, which can be viewed as a Lewis base, in the first step. This reaction leads to formation of a new C–X bond and a new sp3 carbon in a positively charged intermediate called an arenium ion, where X is the electrophile. The positively charged intermediate (the arenium ion) is resonance stabilized, but not aromatic. Loss of a proton from the sp3 carbon that is “adjacent” to the positive carbon in the arenium ion, in what is effectively an E1 process (see Sec. 17.A.ii) is driven by rearomatization of the ring from the arenium ion to give the aromatic substitution product. A proton therefore becomes the leaving group in this overall transformation, where X replaces H. The IUPAC designation for this mechanism is AE + DE. Another mechanism, much less common, consists of the opposite behavior: a leaving group departs before the electrophile arrives. In this case, a substituent (not H) is attached to the aromatic ring, and the substituent is lost prior to incorporation of the electrophile. This mechanism, the SE1 mechanism, corresponds to the SN1 mechanism of nucleophilic substitution. Simultaneous attack and departure mechanisms (corresponding to SN2) are not found at all. An addition–elimination mechanism has been postulated in one case (see Reaction 11-6).
11.A.i. The Arenium Ion Mechanism3
In the arenium ion mechanism, the electrophilic species may be produced in various ways, but when H is replaced by X conversion of the aromatic ring to an arenium ion it is basically the same in all cases. For this reason, most attention in the study of this mechanism centers on the identity of the electrophilic entity and how it is produced.
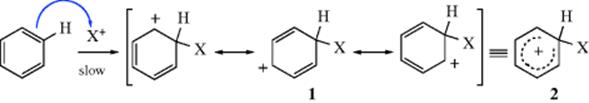
The electrophile may be a positive ion (X+) or be a molecule that has a positive dipole. If it is a positive ion, it is attacked by the ring (a pair of electrons from the aromatic sextet is donated to the electrophile) and the product is a carbocation. This intermediate is a resonance hybrid, as shown in 1, but is often represented as in 2. The H atom to be replaced by X is shown in 1 for convenience. Ions of this type have been called4 Wheland intermediates, σ complexes, but nowadays they are called arenium ions.5 The inherent stability associated with aromaticity is no longer present in 1, but the ion is stabilized by resonance. For this reason, the arenium ion is generally a highly reactive intermediate, although there are cases in which it has been isolated (see below).
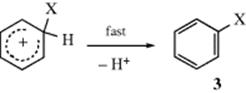
Carbocations can react in various ways (see Sec. 5.A.iii), but for this type of ion the most likely pathway6 is loss of either X+ or H+. In the second step of the mechanism, the reaction proceeds with loss of the proton and the aromatic sextet is restored in the final product (3). The second step is nearly always faster than the first, making the first rate determining, and the reaction is second order. If formation of the attacking species is slower still, the aromatic compound does not take part in the rate expression at all. If X+ is lost, there is no net reaction, but if H+ is lost, an aromatic substitution has taken place and a base (generally the counterion of the electrophilic species although solvents can also serve this purpose) is necessary to help remove it.
If the electrophilic species is not an ion but a molecule with a polarized covalent bond, the product must have a negative charge unless part of the dipole, with its pair of electrons, is broken off somewhere in the process, as in the conversion of 4 to 5. Note that when the aromatic ring attacks X, Z may be lost directly to give 5.
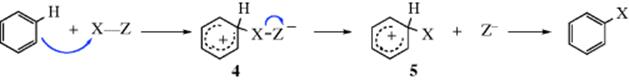
The electrophilic entities and how they are formed are discussed for each reaction in the reactions section of this chapter.
The evidence for the arenium ion mechanism is mainly of two kinds:
1. Isotope Effects. If the hydrogen ion departs before the arrival of the electrophile (SE1 mechanism) or if the arrival and departure are simultaneous, there should be a substantial isotope effect (i.e., deuterated substrates should undergo substitution more slowly than non-deuterated compounds) because, in each case, the C–H bond is broken in the rate-determining step. However, in the arenium ion mechanism, the C–H bond is not broken in the rate-determining step, so no isotope effect should be found. Many such studies have been carried out and, in most cases, especially in the case of nitrations, there is no isotope effect.7 This result is incompatible with either the SE1 or the simultaneous mechanism.
However, in many instances, isotope effects have been found. Since the values are generally much lower than expected for either the SE1 or the simultaneous mechanisms (e.g., 1–3 for kH/kD instead of 6–7), there must be another explanation. For the case where hydrogen is the leaving group, the arenium ion mechanism can be summarized:
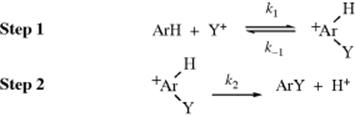
The small isotope effects found most likely arise from the reversibility of step 1 by a partitioning effect.8 The rate at which ArHY+ reverts to ArH should be essentially the same as that at which ArDY+ (or ArTY+) reverts to ArD (or ArT), since the Ar–H bond is not cleaving. However, ArHY+ should go to ArY faster than either ArDY+ or ArTY+, since the Ar–H bond is broken in this step. If k2 ![]() k−1, this does not matter; since a large majority of the intermediates go to product, the rate is determined only by the slow step (k21[ArH][Y+]) and no isotope effect is predicted. However, if k2 ≤ k−1, reversion to starting materials is important. If k2 for ArDY+ (or ArTY+) is <k2for ArHY+, but k−1 is the same, then a larger proportion of ArDY+ reverts to starting compounds. That is, k2/k−1 (the partition factor) for ArDY+ is less than that for ArHY+. Consequently, the reaction is slower for ArD than for ArH and an isotope effect is observed.
k−1, this does not matter; since a large majority of the intermediates go to product, the rate is determined only by the slow step (k21[ArH][Y+]) and no isotope effect is predicted. However, if k2 ≤ k−1, reversion to starting materials is important. If k2 for ArDY+ (or ArTY+) is <k2for ArHY+, but k−1 is the same, then a larger proportion of ArDY+ reverts to starting compounds. That is, k2/k−1 (the partition factor) for ArDY+ is less than that for ArHY+. Consequently, the reaction is slower for ArD than for ArH and an isotope effect is observed.
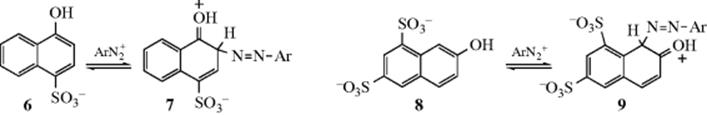
One circumstance that could affect the k2/k−1 ratio is steric hindrance. Thus, diazonium coupling of 6 gave no isotope effect, while coupling of 8 gave a kH/kD ratio of 6.55.9 For steric reasons, it is much more difficult for 9 to lose a proton (it is harder for a base to approach) than it is for 7, so k2 is greater for the latter. Since no base is necessary to remove ArN2+, k−1 does not depend on steric factors10 and is about the same for each. Thus the partition factor k2/k−1 is sufficiently different for 7 and 9 that 8 exhibits a large isotope effect and 6 exhibits none.11 Base catalysis can also affect the partition factor, since an increase in base concentration increases the rate at which the intermediate goes to product without affecting the rate at which it reverts to starting materials. In some cases, isotope effects can be diminished or eliminated by a sufficiently high concentration of base.
Evidence for the arenium ion mechanism has also been obtained from other kinds of isotope-effect experiments, involving substitutions of the type
![]()
where M is Si, Ge, Sn, or Pb, and R is methyl or ethyl. In these reactions, the proton is the electrophile. If the arenium ion mechanism is operating, then the use of D3O+ should give rise to an isotope effect, since the D–O bond would be broken in the rate-determining step. Isotope effects of 1.55–0.05 were obtained,12 in accord with the arenium ion mechanism.
2. Isolation of Arenium Ion Intermediates. Very strong evidence for the arenium ion mechanism comes from the isolation of arenium ions in a number of instances.13 For example, 7 was isolated as a solid with a
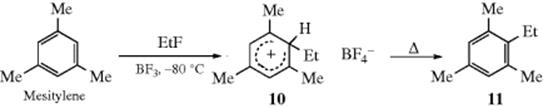
melting point of −15 °C from treatment of mesitylene with ethyl fluoride and the catalyst BF3 at −80 °C. When 10 was heated, the normal substitution product (11) was obtained.14 Even the simplest such ion, the benzenonium ion (12) has been prepared in HF–SbF5–SO2ClF–SO2F2 at −134 °C, where it could be studied
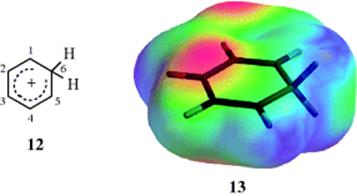
spectrally.15 The ![]() NMR spectra of the benzenonium ion16 and the pentamethylbenzenonium ion17 give graphic evidence for the charge distribution shown in 12 (see the electron density map for the arenium ion, 13). According to this, the 1, 3, and 5 carbons, each of which bears a positive charge of about +1/3 [note that C-1,-3,-5 (numbering from 12) are lighter, indicating less electron density in 13, whereas C-2,-4 are darker for higher electron density], should have a greater chemical shift in the NMR than the 2 and 4 carbon atoms, which are uncharged. The spectra bear this out. For example,
NMR spectra of the benzenonium ion16 and the pentamethylbenzenonium ion17 give graphic evidence for the charge distribution shown in 12 (see the electron density map for the arenium ion, 13). According to this, the 1, 3, and 5 carbons, each of which bears a positive charge of about +1/3 [note that C-1,-3,-5 (numbering from 12) are lighter, indicating less electron density in 13, whereas C-2,-4 are darker for higher electron density], should have a greater chemical shift in the NMR than the 2 and 4 carbon atoms, which are uncharged. The spectra bear this out. For example, ![]() NMR chemical shifts for 12 are C-3: 178.1; C-1 and C-5: 186.6; C-2 and C-4: 136.9, and C-6: 52.2.16
NMR chemical shifts for 12 are C-3: 178.1; C-1 and C-5: 186.6; C-2 and C-4: 136.9, and C-6: 52.2.16
In Chapter 3, it was mentioned that positive ions can form addition complexes with π systems. Since the initial step of electrophilic substitution involves attack of a positive ion by an aromatic ring, it has been suggested18 that such a complex, called a π-complex (represented as 14), is formed first, and then is converted to the arenium ion (15).19 Stable solutions of arenium ions or π complexes (e.g., with Br2, I2, picric acid, Ag+, or HCl) can be formed.20For example, π-complexes are formed when aromatic hydrocarbons are treated with HCl alone, but the use of HCl plus a Lewis acid (e.g., AlCl3) gives arenium ions. The two types of solution have very different properties. For example, a solution of an arenium ion is colored and conducts electricity, which shows that positive and negative ions are present, while a π complex formed from HCl and benzene is colorless and does not conduct a current. Furthermore, when DCl is used to form a π complex, no deuterium exchange takes place (because there is no covalent bond between the electrophile and the ring), while formation of an arenium ion with DCl and AlCl3 gives deuterium exchange. The relative stabilities of some methylated arenium ions and π complexes are shown in Table 11.1. The arenium ion stabilities listed were determined by the relative basicity of the substrate toward HF.21 The π-complex stabilities are relative equilibrium constants for the reaction22 between the aromatic hydrocarbon and HCl. As shown in Table 11.1, the relative stabilities of the two types of species are very different: The π complex stability changes very little with methyl substitution, but the arenium ion stability changes a great deal. Note that stable arenium ions have been obtained from large methylene-bridged polycyclic aromatic hydrocarbons.23
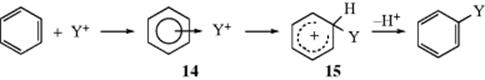
How can we tell if 14 is present on the reaction path? If it is present, there are two possibilities: (1) The formation of 14 is rate determining (the conversion of 14 to 15 is much faster), or (2) the formation of 14 is rapid, and the conversion 14 to 15 is rate determining. One way to ascertain which species is formed in the rate-determining step in a given reaction is to use the stability information given in Table 11.1. We measure the relative rates of reaction of a given electrophile with the series of compounds listed in Table 11.1. If the relative rates resemble the arenium ion stabilities, we conclude that the arenium ion is formed in the slow step; but if they resemble the stabilities of the π complexes, the latter are formed in the slow step.24 When such experiments are carried out, it is found in most cases that the relative rates are similar to the arenium ion and not to the π complex stabilities. For example, Table 11.1lists chlorination rates.22 Similar results were obtained in room temperature bromination with Br2 in acetic acid25 and in acetylation with CH3CO+ SbF6−.26 It is clear that in these cases the π complex either does not form at all, or if it does, its formation is not rate determining (unfortunately, it is very difficult to distinguish between these two possibilities).
Table 11.1 Relative Stabilities of Arenium Ions, π-Complexes, and Relative Rates of Chlorination and Nitrationa
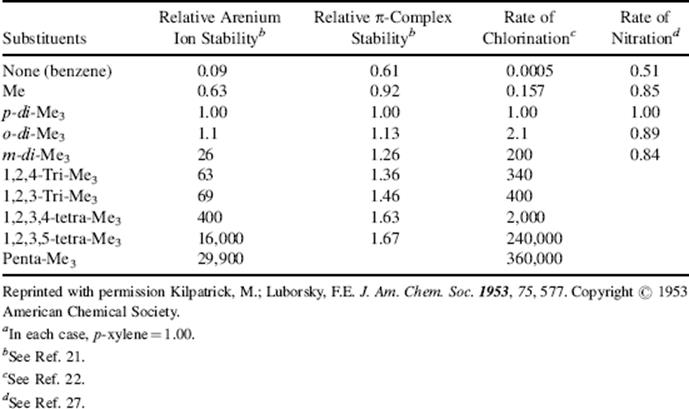
On the other hand, in nitration with the powerful electrophile NO2+ (in the form of NO2+ BF4−), the relative rates resembled π complex stabilities much more than arenium ion stabilities (Table 11.1).27 Similar results were obtained for bromination with Br2 and FeCl3 in nitromethane. These results were taken to mean28 that in these cases π complex formation is rate determining. However, graphical analysis of the NO2+ data showed that a straight line could not be drawn when the nitration rate was plotted against π-complex stability,29 which casts doubt on the rate-determining formation of a π complex in this case.30 There is other evidence, from positional selectivities (discussed in Sec. 11.D), that some intermediate is present before the arenium ion is formed, whose formation can be rate determining with powerful electrophiles. Not much is known about this intermediate, which is given the nondescriptive name encounter complex and generally depicted as 16. The arenium complex mechanism is therefore written as:31
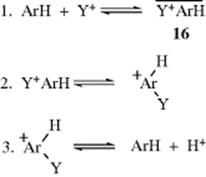
For the reason given above and for other reasons, it is unlikely that the encounter complex is a π complex, but just what kind of attraction exists between Y+ and ArH is not known, other than the presumption that they are together within a solvent cage (see also, Sec. 11.D). There is evidence (from isomerizations occurring in the alkyl group, as well as other observations) that π complexes are present on the pathway from substrate to arenium ion in the gas-phase protonation of alkylbenzenes.32
11.A.ii. The SE1 Mechanism
The SE1 mechanism (substitution electrophilic unimolecular) is rare, being found only in certain cases in which carbon is the leaving atom (see Reactions 11-33 and 11-35) or when a very strong base is present (see Reactions 11-1, 11-10, and 11-39).33 It consists of two steps with an intermediate carbanion. The IUPAC designation is DE + AE.
![]()
Reactions 12-41, 12-45, and 12-46 also take place by this mechanism when applied to aryl substrates.