March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 11. Aromatic Substitution, Electrophilic
11.B. Orientation and Reactivity
11.B.i. Orientation and Reactivity in Monosubstituted Benzene Rings34
When an electrophilic substitution reaction is performed on a monosubstituted benzene, the new group may be directed primarily to the ortho, meta, or para position and the substitution may be slower or faster than with benzene itself.35 The group already on the ring determines which position the new group will take and whether the reaction will be slower or faster than with benzene. Groups that increase the reaction rate are called activating and those that slow it are deactivating. Some groups are predominantly meta directing; all of these are deactivating. Others are mostly ortho–para directing; while some of these (e.g., halogens) are deactivating too, most are activating. Groups direct predominantly, but usually not exclusively. For example, nitration of nitrobenzene gave 93% m-dinitrobenzene, 6% of the ortho, and 1% of the para isomer.
The orientation and reactivity effects are explained on the basis of resonance and field effects of each group on the stability of the intermediate arenium ion. To understand why we can use this approach, it is necessary to know that in these reactions the product is usually kinetically and not thermodynamically controlled (see Sec. 6.F). Some of the reactions are irreversible and the others are usually stopped well before equilibrium is reached. Therefore, which of the three possible intermediates is formed is dependent not on the thermodynamic stability of the products, but on the activation energy necessary to form each of the three intermediates. It is not easy to predict which of the three activation energies is lowest, but it is necessary to make the assumption that the free energy profile resembles either Fig. 6.2a or b. In either case, the transition state is closer in energy to the arenium ion intermediate than to the starting compounds. Invoking the Hammond postulate (Sec. 6.G), assume that the geometry of the transition state also resembles that of the intermediate and that anything that increases the stability of the intermediate will also lower the activation energy necessary to attain it. Since the intermediate, once formed, is rapidly converted to products, the relative stabilities of the three intermediates can be used as guides to predict which products will predominantly form. Of course, if reversible reactions are allowed to proceed to equilibrium, product ratios that are quite different may be obtained. For example, the sulfonation of naphthalene at 80 °C, where the reaction does not reach equilibrium, gives mostly α-naphthalenesulfonic acid,36 while at 160 °C, where equilibrium is attained, the β isomer predominates37 (the α isomer is thermodynamically less stable because of steric interaction between the SO3H group and the hydrogen at the 8 position).
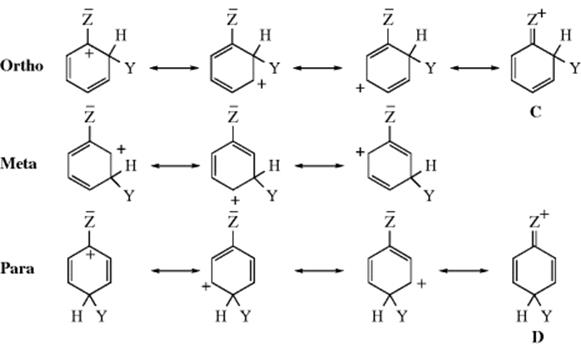
The three possible ions from incorporation of Y at the ortho, meta, and para positions are shown below, and each arenium ion obviously has a positive charge in the ring. It is therefore possible to predict that any group Z that has an electron-donating field effect (+I, Z will have a − charge or a δ− dipole in most cases) should stabilize all three ions (relative to 1), since electron donation to a positive center is stabilizing. On the other hand, electron-withdrawing groups (−I, Z will have a + charge or a δ+ dipole in most cases) will increase the positive charge on the ring (like charges repel), and destabilize the arenium ion. Formation of a stabilized ion should be faster than benzene, which generates 1, or activating, but formation of a destabilized ion should be slower, or deactivating. Such field effects should taper off with distance and are thus strongest at the carbon connected to the group Z (known as the ipso carbon). Of the three arenium ions, only the ortho and para have any positive charge at this carbon. None of the canonical forms of the meta ion has a positive charge at the ipso carbon. Therefore, +I groups should stabilize all three ions, but mostly the ortho and para, so they should be not only activating but ortho–para-directing as well. On the other hand, −I groups, by removing electron density, should destabilize all three ions, but mostly the ortho and para, and should be not only deactivating but also meta directing.

These conclusions are correct as far as they go, but they do not lead to the proper results in all cases. In many cases, there is resonance interaction between Z and the ring; this also affects the relative stability, in some cases in the same direction as the field effect, in others differently.
Some substituents have a pair of electrons (usually unshared) that may be contributed toward the ring. Two of the three (ortho and para) arenium ions would then have a fourth resonance contributor as shown. For each ion, the same three canonical forms can be drawn as before, but now we can draw an extra form for the ortho and para ions. The stability of these two ions is increased by the extra form not only because it is another canonical form, but also because it is more stable than the others and makes a greater contribution to the hybrid. Every atom (except, of course, hydrogen) in these forms (C and D) has a complete octet, while all the other forms have one carbon atom with a sextet. No corresponding form can be drawn for the meta isomer. The inclusion of this form in the hybrid lowers the energy not only because of rule 6 (Sec. 2.E), but also because it spreads the positive charge over a larger area, out onto group Z. Groups with a pair of electrons (e.g., the halogens) to contribute would be expected, then, in the absence of field effects, not only to direct ortho and para, but also to activate these positions for electrophilic attack.
On the basis of these discussions, we can distinguish three types of groups.
1. Groups that Contain an Unshared Pair of Electrons on the Atom Connected to the Ring. In this category are O−, NR2, NHR, NH2,38 OH, OR, NHCOR, OCOR, SR, and the four halogens.39 The halogens deactivate the aromatic ring to substitution (the rate of reaction is slower than that of benzene), and this effect may arise from the unique energy level of the halogen lone-pair orbital, which is higher than the adjacent π molecular orbital of benzene (π1).40 The widely held explanation for this, however, is that the halogens have a −I effect. The SH group would probably belong here too, except that in the case of thiophenols, electrophiles usually attack the sulfur rather than the ring, and ring substitution is not feasible with these substrates.41 The resonance explanation predicts that all these groups should be ortho–para directing, and they are, though all except O− are electron withdrawing by the field effect (Sec. 1.I). Therefore, for these groups, resonance is more important than the field effect. This result is especially true for NR2, NHR, NH2, and OH, which are strongly activating, as is O−. The other groups are mildly activating, except for the halogens, which are deactivating. Fluorine42 is the least deactivating, and fluorobenzenes usually show a reactivity approximating that of benzene itself. The other three halogens deactivate about equally. In order to explain why chlorine, bromine, and iodine deactivate the ring, even though they direct ortho–para, assume that the canonical forms C and D make such great contributions to the respective hybrids that they make the ortho and para arenium ions more stable than the meta, even though the −I effect of the halogen is withdrawing sufficient electron density from the ring to deactivate it. The three halogens make the ortho and para ions more stable than the meta, but less stable than the unsubstituted arenium ion (1). For the other groups that contain an unshared pair, the ortho and para ions are more stable than either the meta or the unsubstituted ion. For most of the groups in this category, the meta ion is more stable than 1, so that groups, such as NH2 and OH, activate the meta positions too, but not as much as the ortho and para positions (see also, the discussion in Sec. 11.C).
2. Groups that Lack an Unshared Pair on the Atom Connected to the Ring and that Are −I. In this category are, in approximate order of decreasing deactivating ability, NR3+, NO2, CF3,43 CN, SO3H, CHO, COR, CO2H, CO2R, CONH2, CCl3, and NH3+. Also in this category are all other groups with a positive charge on the atom directly connected to the ring44 (SR2+, PR3+, etc.) and many groups with positive charges on atoms farther away, since often these are still powerful −I groups. The field-effect explanation predicts that these should all be meta directing and deactivating, and (except for NH3+) this is the case. The NH3+ group is an anomaly, since this group directs para about as much as or a little more than it directs meta.45 The NH2Me+, NHMe2+, and NMe3+ groups all give more meta than para substitution, the percentage of para product decreasing with the increasing number of methyl groups.46
3. Groups that Lack an Unshared pair on the Atom connected to the Ring and that Are ortho–para Directing. In this category are alkyl groups,47 aryl groups, and the COO− group,48 all of which activate the ring. Since aryl groups are −I groups, they might seem to belong to category 2. They are nevertheless ortho–para directing and activating. This can be explained in a similar manner as in category 1, with a pair of electrons from the aromatic sextet playing the part played by the unshared pair, so that forms like E are generated The effect of negatively charged groups like CO2− is easily explained by the field effect (negatively charged groups are of course electron donating), since there is no resonance interaction between the group and the ring. The effect of alkyl groups can be explained in the same way, but, in addition, canonical forms can be drawn, even though there is no unshared pair. These, of course, are hyperconjugation forms like F (see above). This effect, like the field effect, predicts activation and ortho–para direction, so that it is not possible to say how much each effect contributes to the result. Another way of looking at the effect of alkyl groups, which sums up both field and hyperconjugation effects, is that (for Z = R) the ortho and para arenium ions are more stable because each contains a form (A and B) that is a tertiary carbocation, while all the canonical forms for the meta ion and for 1 are secondary carbocations. In activating ability, alkyl groups usually follow the Baker–Nathan order (Sec. 2.M), but not always.49
11.B.ii. The Ortho/Para Ratio50
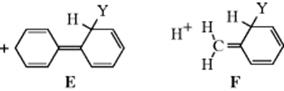
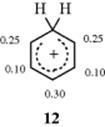
When an ortho–para directing group is on a ring, it is usually difficult to predict how much of the product will be the ortho isomer and how much is the para isomer. Indeed, these proportions can depend greatly on the reaction conditions. For example, chlorination of toluene gives an ortho/para ratio anywhere from 62:38 to 34:66.51 Nevertheless, certain points can be made. On a purely statistical basis there would be 67% ortho and 33% para, since there are two ortho positions and only one para. However, the phenonium ion (12), which arises from protonation of benzene, has the approximate charge distribution shown52 (see 13 as well). If this model were accepted for the arenium ion in aromatic substitution, a para substituent would have a greater stabilizing effect on the adjacent carbon than an ortho substituent. If other effects are absent, this would mean that >33% para and <67% ortho substitution would be found. In hydrogen exchange (Reaction 11-1), where other effects are absent, it has been found for a number of substituents that the average ratio of the logarithms of the partial rate factors for these positions (see Sec. 11.C for a definition of partial rate factor) was close to 0.865,53 which is not far from the value predicted from the ratio of charge densities in 12. This picture is further supported by the fact that meta-directing groups, which destabilize a positive charge, give ortho/para ratios >67:3354 (of course, the total amount of ortho and para substitution with these groups is small, but the ratios are generally >67:33). Another important factor is the steric effect. If either the group on the attacking ring or the group on the electrophile is large, steric hindrance inhibits formation of the ortho product and increases the amount of the para isomer. An example may be seen in the nitration, under the same conditions, of toluene and tert-butylbenzene. The former gave 58% of the ortho compound and 37% of the para, while the more bulky tert-butyl group gave 16% of the ortho product and 73% of the para.55 Some groups are so large that they direct almost entirely para.
When the ortho–para directing group is one with an unshared pair (this of course applies to most of them), there is another effect that increases the amount of para product at the expense of the ortho. A comparison of the intermediates involved (see above) shows that C is a canonical form with an ortho-quinoid structure, while D has a para-quinoid structure. para-Quinones are more stable than the ortho isomers, so it seems reasonable to assume that D is more stable than C, and therefore contributes more to the hybrid and increases its stability compared to the ortho intermediate.
It has been shown that it is possible to compel regiospecific para substitution by enclosing the substrate molecules in a cavity from which only the para position projects. Anisole was chlorinated in solutions containing a cyclodextrin, a molecule in which the anisole is almost entirely enclosed (see Fig. 3.4). With a high enough concentration of cyclodextrin, it was possible to achieve a para/ortho ratio of 21.656 (in the absence of the cyclodextrin the ratio was only 1.48). This behavior is a model for the regioselectivity found in the action of enzymes.
11.B.iii. Ipso Attack
Orientation has been discussed previously in the case of monosubstituted benzenes entirely in terms of attachment at the ortho, meta, and para positions, but attachment at the position bearing the substituent (called the ipso position57) can also be important. Ipso attack has mostly been studied for nitration.58 When attack of NO2+ leads to incorporation at the ipso position there are at least five possible fates for the resulting arenium ion (17).
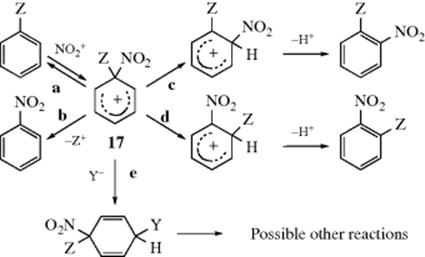
Path a. The arenium ion can lose NO2+ and revert to the starting compounds. This results in no net reaction and is often undetectable.
Path b. The arenium ion can lose Z+, in which case this is simply aromatic substitution with a leaving group other than H (see Reactions 11-33–11-41).
Path c. The electrophilic group (in this case NO2+) can undergo a 1,2-migration, followed by loss of the proton. The product in this case is the same as that obtained by direct attachment of NO2+ at the ortho position of PhZ. It is not always easy to tell how much of the ortho product in any individual case arises from this pathway,59 though there is evidence that it can be a considerable proportion. Because of this possibility, many of the reported conclusions about the relative reactivity of the ortho, meta, and para positions are cast into doubt, since some of the product may have arisen not from direct attachment at the ortho position, but from attachment at the ipso position followed by rearrangement.60
Path d. The ipso substituent (Z) can undergo 1,2-migration, which also produces the ortho product (although the rearrangement would become apparent if there were other substituents present). The evidence is that this pathway is very minor, at least when the electrophile is NO2+.61
Path e. Attack of a nucleophile on 17. In some cases, the products of such an attack (cyclohexadienes) have been isolated62 (this is 1,4-addition to the aromatic ring), but further reactions are also possible.
11.B.iv. Orientation in Benzene Rings with More Than One Substituent63
It is often possible in these cases to predict the correct isomer. In many cases, the groups already on the ring reinforce each other. Thus, 1,3-dimethylbenzene is substituted at the 4 position (ortho to one group and para to the other), but not at the 5 position (meta to both). Likewise, the incoming group in p-chlorobenzoic acid goes to the position ortho to the chloro and meta to the carboxyl group.
When the groups oppose each other, predictions may be more difficult, as in N-acetyl-2-methoxyaniline. In a case such as where two groups of about equal directing ability are in competing positions, all four products can be expected, and it is not easy to predict the proportions, except that steric hindrance should probably reduce the yield of substitution ortho to the acetamido group, especially for large electrophiles. Mixtures of about equal proportions are frequent in such cases. Nevertheless, even when groups on a ring oppose each other, there are some regularities.
1. If a strong activating group competes with a weaker one or with a deactivating group, the former controls. Thus o-cresol gives substitution mainly ortho and para to the hydroxyl group and not to the methyl. For this purpose, we can arrange the groups in the following order: NH2, OH, NR2, O− > OR, OCOR, NHCOR > R, Ar > halogen > meta-directing groups.
2. All other things being equal, a third group is least likely to enter between two groups in the meta relationship. This is the result of steric hindrance and increases in importance with the size of the groups on the ring and with the size of the attacking species.64
3. When a meta-directing group is meta to an ortho–para directing group, the incoming group primarily goes ortho to the meta-directing group rather than para. For example, chlorination of 18 gives mostly 19. The importance of this effect is underscored by the fact that 20, which is in violation of the preceding rule, is formed in smaller amounts, but 21 is not formed at all. This is called the ortho effect,65 and many such examples are known.66Another is the nitration of p-bromotoluene, which gives 2,3-dinitro-4-bromotoluene. In this case, once the first nitro group came in, the second was directed ortho to it rather than para, even though this means that the group has to come in between two groups in the meta position. There is no good explanation yet for the ortho effect, though possibly there is intramolecular assistance from the meta-directing group.
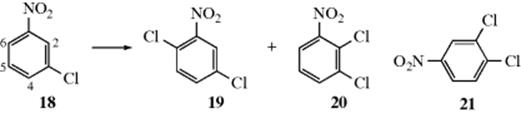
It is interesting that chlorination of 18 illustrates all three rules. Of the four positions open to the electrophile, the 5 position violates rule 1, the 2 position rule 2, and the 4 position rule 3. The principal attachment is therefore at position 6.
11.B.v. Orientation in Other Ring Systems67
In fused ring systems, the positions are not equivalent and there is usually a preferred orientation, even in the unsubstituted hydrocarbon. The preferred positions may often be predicted as for benzene rings. Thus it is possible to draw more canonical forms for the arenium ion when attack by naphthalene leads to attachment of the electrophile at the α position than when attack by naphthalene leads to attachment of the electrophile at the β position. Therefore, the α position is the preferred site of attachment,68 although, as previously mentioned (Sec. 11.B.i), the isomer formed by substitution at the β-position is thermodynamically more stable and is the product if the reaction is reversible and equilibrium is reached. Because of the more extensive delocalization of charges in the corresponding arenium ions, naphthalene is more reactive than benzene and substitution is faster at both positions. Similarly, anthracene, phenanthrene, and other fused polycyclic aromatic hydrocarbons are also substituted faster than benzene.
Heterocyclic compounds, too, have nonequivalent positions, and the principles are similar,69 in terms of mechanism, and rate data is available.70 Furan, thiophene, and pyrrole are chiefly substituted at the 2 position, and all are substituted faster than benzene.71 Pyrrole is particularly reactive, with a reactivity approximating that of aniline or the phenoxide ion. For pyridine,72 it is not the free base that must attack the electrophile but the conjugate acid (the pyridinium ion),73 making the reactivity much less than that of benzene, being similar to that of nitrobenzene. The 3 position is most reactive in electrophilic substitution reactions of pyridine. However, groups can be introduced into the 4 position of a pyridine ring indirectly, by performing the reaction on the corresponding pyridine N-oxide.74 Note that calculations show that the 2-pyridyl and 2-pyrimidyl cations are best represented as ortho-hetarynium ions, being more stable than their positional, nonconjugated isomers by as much as 18–28 kcal mol−1 (75–117 kJ mol−1).75
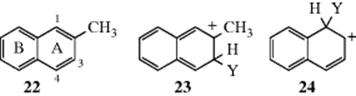
When fused ring systems contain substituents, successful predictions can often be made by using a combination of the above principles. Thus, ring A of 2-methylnaphthalene (22) is activated by the methyl group; ring B is not (although the presence of a substituent in a fused ring system affects all the rings,76 the effect is generally greatest on the ring to which it is attached). Substitution is therefore expected in ring A. The methyl group activates positions 1 and 3, which are ortho to itself, but not position 4, which is meta to it. However, substitution at the 3 position gives rise to an arenium ion for which it is impossible to write a low-energy canonical form in which ring B has a complete sextet. Only forms like 23 are possible, in which the sextet is no longer intact. In contrast, substitution at the 1 position gives rise to a more stable arenium ion, for which two canonical forms (one of them is 24) can be written in which ring B is benzenoid. We thus predict predominant substitution at C-1, and that is what is generally found.77 However, in some cases predictions are much harder to make. For example, chlorination or nitration of 25gives mainly the 4 derivative, but bromination yields chiefly the 6 compound.78
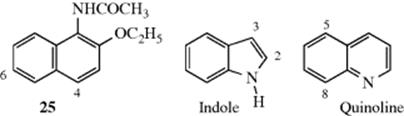
For fused heterocyclic systems too, predictions can be made based on the above principles, although many exceptions are known. Thus, indole is chiefly substituted in the pyrrole ring (at position 3) and reacts faster than benzene, while quinoline generally reacts in the benzene ring, at the 5 and 8 positions, and slower than benzene, though faster than pyridine.
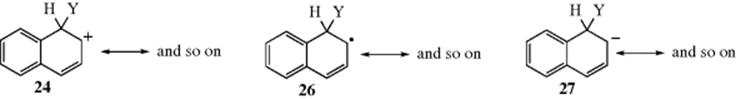
In alternant hydrocarbons (Sec. 2.J), the reactivity at a given position is similar for electrophilic, nucleophilic, and free radical substitution, because the same kind of resonance can be shown in all three types of intermediate (cf. 24, 26, and 27). Attachment of the electrophile at the position that will best delocalize a positive charge will also best delocalize a negative charge or an unpaired electron. Most results are in accord with these predictions. For example, naphthalene is attacked primarily at the 1 position by NO2+, NH2−, and Ph√, and always more readily than benzene.
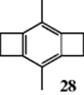
When strain due to a ring fused on an aromatic ring deforms that ring out of planarity, the molecule is more reactive to electrophilic aromatic substitution.79 This has been explained by the presence of a shortened bond for the sp2hybridized carbon, increasing the strain at that position. This finding is known as the Mills–Nixon effect.80 There is EPR evidence (see Sec. 5.C.i) for 3,6-dimethyl-1,2,4,5-tetrahydrobenzo-bis-cyclobutene (28), which supports the Mills–Nixon effect,81 and a theoretical study, which supports this.82 However, ab initio studies of triannelated benzene rings shows no evidence for the Mills–Nixon effect, and a new motif for bond-alternating benzenes was proposed.83 Indeed, it is argued that the Mills–Nixon effect is not real.84