CHEMICAL BIOLOGY
Mathematical Modeling of Biological Signaling Networks
Jason M. Haugh, North Carolina State University, Raleigh, North Carolina
doi: 10.1002/9780470048672.wecb646
Intracellular signaling networks, which are composed of interconnected biochemical pathways, regulate and actuate responses such as cell-cycle progression and cell migration, survival, and differentiation. Although our knowledge of the intricate biochemical mechanisms at the level of individual proteins and molecular interactions is ever expanding, those details leave us with an even murkier view of how the complex network operates as a whole. True understanding requires knowing not only what happens at the molecular level but also how these mechanisms influence the precise magnitude, timing, and spatial localization of signal transduction processes. Hence, mathematical modeling and analysis has emerged in recent years as a legitimate approach for interpreting experimental results and generating novel hypotheses for additional study and model refinement. Once conducted in isolation and scorned by most biologists, quantitative modeling has moved into the mainstream as a powerful tool for the analysis of cell signaling. In this article, the biological, chemical, and physical underpinnings of this approach are presented, as are its current applications and future challenges.
Biological research has been focused increasingly on the basis for cell regulation and function at the molecular level, and as a result, we now have a detailed map of how intracellular molecules are organized to form signal transduction pathways and cascades (Fig. 1). The “protein jungle,” as articulated by Bray a decade ago (1), is characterized by a wealth of qualitative information about the connectivity of pathways (the so-called interactome) but relatively little in the way of quantitative measurements. For example, one might wish to know how some meaningful quantity, such as the activity of a particular enzyme, changes as a function of time and stimulus in a given cell type. A quantitative assay could be developed for measuring that quantity. Understanding those kinetics fully, however, would require detailed knowledge of the underlying regulation mechanisms, of which several typically exist. Reconstituting those mechanisms in a test tube, in most cases, is prohibitively difficult, as is their systematic manipulation in the cell. Ultimately, the regulatory mechanisms would have to be characterized in terms of equilibrium and rate constants and intracellular concentrations. Finally, it would be useful to develop a correlation between the magnitude and timing of that pathway and the quality of cellular responses, such as rates of proliferation and probability of survival. Even if obtaining such a data set were feasible, it is not obvious how one would analyze it and extract useful insights and predictions from it.
This article deals with the mathematical modeling of signal transduction pathways and networks, which has emerged as a powerful tool that can aid in interpreting such quantitative data sets. In principle, quantitative models offer three advantages over the conceptual “arrow diagrams” that are encountered routinely in the signaling literature. First, a consequence of their mathematical construction is that they are precise, and the inherent assumptions may be laid out clearly. Second, to the extent that the underlying molecular biology is known, quantitative models can be mechanistic. Mechanistic models are based on established physico-chemical principles, in which case the form of the model equations is determined to a significant extent by the hypothetical mechanisms assumed. Thus, it is possible to evaluate the relative merits of different candidate mechanisms by comparison with experiment. In contrast with mechanistic models, phenomenological models are meant only to capture experimentally observed relationships in an empirical way. They naturally are less powerful, but they serve a definite and useful role and are appropriate in situations in which the underlying mechanisms are less certain. A prominent example is the sort of statistical, correlative models central to the field of bioinformatics; although such approaches are not discussed in detail here, they have been applied with great success in the analysis of signal transduction networks (2, 3). Third, a model that has been validated against an appropriate amount of quantitative data may be used to design and predict the outcomes of novel experiments and generate new hypotheses. Evaluating those outcomes by experiment inevitably reveals inconsistencies in the model, and hence the model must be refined in an iterative manner. This is in essence the same process by which qualitative, conceptual models are refined over time, although one could argue that quantitative models are far more sensitive to hypothesis testing because of their precise nature.

Figure 1. Partial representation of a signal transduction network, mediated by a cell surface receptor. The molecules are organized hierarchically (roughly, from top to bottom) according to their functions as adaptors, receptor-recruited enzymes, membrane-associated substrates, and effector kinases. Arrows represent activation mechanisms, whether by complex formation or covalent modification; 'T' bars indicate negative regulation.
Signal Transduction Networks: Biological Background
The molecular basis of signal transduction lies in the noncovalent interactions among cell-associated proteins, lipids, and other biomolecules, which leads to the formation of transient complexes, and the covalent modifications of those biomolecules by specific, enzyme-catalyzed reactions. Enzymatic activities, in turn, are regulated by interactions with other proteins, lipids, or small molecules/ions and by covalent modifications. Mediating all of these processes are receptors, cell-associated proteins responsible for sensing specific chemical factors (ligands), typically through noncovalent interactions at the cell surface. Receptors activated by ligation, or in some cases by mechanical forces, actuate intracellular pathways and thus preside over signal transduction.
Receptor-ligand interactions and receptor dynamics
Receptors are responsible for transmitting information about the external environment to the cell internum, and therefore their mechanism of activation and action must be well characterized before the details of the intracellular networks can be analyzed and modeled mathematically. In most cases, receptor activation is induced by the noncovalent binding of a specific ligand. Expression of the cognate receptor, typically existing at copy numbers of 102-105 per cell, is tantamount to the ability of a cell to respond to the ligand, and thus chemical ligands signal specific cell types. Most ligands are soluble and diffusible, and such ligands are termed growth factors, cytokines, hormones, or agonists depending on the type of receptor engaged and/or the response elicited. Other ligands are associated with the extracellular matrix or other cells and mediate cell-matrix adhesion and cell-cell interactions, respectively, in addition to signaling functions. In many cases, ligation induces receptor dimerization or oligomerization as a prerequisite for signal transduction. For example, it is well established that receptor tyrosine kinases (RTKs), receptors that engage certain growth factors in a variety of cell types, must dimerize for their intrinsic enzymatic activity to phosphorylate specific tyrosine residues in the cytoplasmic portion of each receptor. The decoration of the receptor with phospho-tyrosine provides a scaffold for the recruitment of a host of signaling enzymes (4).
Receptors and receptor-ligand complexes on the cell surface are not static. Their lateral mobility in the plasma membrane enables receptor dimerization, as described above, but another critical aspect of receptor dynamics is receptor trafficking, whereby components of the plasma membrane are internalized, delivered to intracellular compartments called endosomes, and sorted either for recycling back to the cell surface or for enzymatic degradation (5). Thus, differential sorting of activated and inactive receptors provides a mechanism for regulating the number of cell surface receptors in response to chronic ligand exposure, a process called receptor downregulation, and can contribute to the clearance of external ligands over time. Typically, activated receptors are internalized at a faster rate and their intracellular sorting fate depends on the persistence of the receptor-ligand interaction in endosomes. Any realistic model of receptor-mediated signal transduction should take into consideration the processes of ligand binding, receptor dimerization or oligomerization, and receptor/ligand trafficking. Receptors that are well characterized, such as the epidermal growth factor receptor, therefore have attracted the most attention from modelers (6).
Modular functions of signaling proteins
Signaling proteins form complexes with other molecules using conserved motifs or domains, which are modular in the sense that the domain by itself typically is sufficient for its function (7, 8). Protein-protein interaction domains include those responsible for binding to phosphorylated receptors and other tyrosine-phosphorylated proteins (Src homology 2 and phospho-tyrosine binding domains) or to proline-rich protein sequences (Src homology 3 domains); other domains are responsible for interactions with lipids (pleckstrin homology, FYVE, C1, and C2 domains). Together, these domains mediate the formation of multimolecular complexes that regulate the activities and mediate the targeting of signaling enzymes. Many signaling enzymes have a handful of such domains, whereas other signaling proteins called adaptors have no enzymatic function but possess domains that mediate the binding of enzymes to signaling complexes. From the standpoint of modeling, this complexity presents a definite challenge.
Physical organization and compartmentalization of signaling processes
Cells are not simply well-mixed reaction vessels, and cell structure plays a critical role in signal transduction. Besides its obvious barrier function, the physical properties of the plasma membrane and other cell membranes are particularly important. In the inner leaflet of the plasma membrane, certain lipids such as phosphatidylinositols and phosphatidylcholine are substrates for signaling enzymes and can be hydrolyzed to form soluble and membrane-associated products or phosphorylated to yield other lipids with second messenger functions. In signal transduction pathways that do not involve lipid modification reactions, typically a lipid-tethered protein is involved, such as small GTPases of the Ras and Rho families. Given the near ubiquity of biochemical reactions and interactions that occur at cell membranes, it is clear that aspects of both surface and solution chemistry should be considered when formulating models of signal transduction.
In modeling interactions at cell membranes, it is important to consider the mobility of the membrane-associated molecules, which is affected by the fluidity of the membrane bilayer. Often it is assumed that the membrane is isotropic and homogeneous in that respect, at least macroscopically; however, it has long been appreciated that the plasma membrane is organized into ordered and disordered subcompartments (9). Low-density microdomains, including lipid rafts and caveolae, have been found to be enriched with certain receptors, lipids, and lipid-tethered proteins that are involve in signaling, but their role in facilitating and/or regulating signal transduction remains controversial (10, 11). Even in the bulk, disordered plasma membrane, single-molecule imaging has revealed complex molecular mobility dynamics consistent with “hop” diffusion across corral-like barriers (12). These and other aspects of cell membrane organization increasingly have brought signal transduction into the realm of biophysics.
Beyond the distinctions between signaling processes that occur at the plasma membrane and in the cell cytoplasm, certain other intracellular compartments may need to be considered. As a prominent example, the signaling competency of internalized receptors has been studied for 2 decades (13, 14). Some receptor-ligand complexes remain active in endosomes and signal through certain pathways but not others, whereas other complexes dissociate and cease to signal after the delivery to the endosomes. Other internal compartments, such as the Golgi and endoplasmic reticulum, have been found more recently to serve as platforms for certain signaling interactions (15).
Signal Transduction Models: Chemical/Physical Foundations
The premise of molecular biology is that cellular processes are governed by physico-chemical principles, and accordingly, those principles may be used to translate known or hypothetical molecular mechanisms to mathematical equations. In this section, the general principles of chemical kinetics, mass transport, and fluid mechanics used to model chemical reaction systems are reviewed briefly.
Reaction and mass-action kinetics
In chemical kinetics, each molecule type in the system is considered a distinct species and a conservation equation is formulated to account for the change in the amount of each species per unit time. In this context, the system could be a single cell and the species could be cell-associated molecules; different states of a molecule, the simplest distinction being a two-state model (e.g., active versus inactive or phosphorylated versus unphos-phorylated), are considered distinct species as are complexes of multiple molecules. The rate of each biochemical reaction or interaction is expressed in terms of the concentrations of the reacting/interacting species, defining the so-called rate law, which appears as a positive or negative term in the corresponding conservation equations depending on whether the transition generates or consumes the conserved species.
In formulating rate laws, it is common to invoke the law of mass action (Fig. 2a). In the biochemical context, this principle applies to only two types of processes: 1) the union of two species (complex formation), the rate defined as the product of both of their concentrations and a bimolecular rate constant, and 2) a spontaneous, unimolecular transition, such as the dissociation of two species from a complex or the covalent modification of a substrate in a complex with an enzyme, which occurs with a constant mean probability per unit time that defines its rate constant. A rate process that follows mass-action kinetics often is termed an elementary reaction, although that definition technically implies specific criteria that generally are not met for reactions in liquids much less for biomolecules. The advantage of mass action kinetics is that the mathematical form of the rate law is dictated by the mechanism, in which case one needs to specify only the values of the corresponding rate constants. For complicated rate processes, a notable example being the action of highly cooperative enzymes, a more abstract rate law that does not reflect the precise mechanism but nonetheless is in quantitative agreement with experimental measurements might be assumed.

Figure 2. Mathematical modeling of cellular processes: an illustrative example. A. Consider the following mechanism for ligand/receptor dynamics: Cell surface receptors (R) are synthesized at a constant rate and bind reversibly with a ligand (L) to form a complex (C). Both free and bound receptors are internalized and later degraded, but they do so at different rates. Based on this mechanism, the law of mass action is used to construct the conservation equations for R and C. B. Addition of transport effects. In the case of autocrine signaling, the cell is both the source of the secreted ligand and the responder. Spherical geometry is adopted, and a simple reaction/diffusion model is used to calculate the ligand concentration profile at steady state, assuming a dilute suspension of cells and the same receptor dynamics as in A. This profile is given by [L] = [L]S(a/r), where [L]S is the value of [I] at the cell surface (r = a). It is shown readily that the maximum value of [L]s, achieved when no receptor binding exists, is equal to aVL / DL, where VL is the cell rate of ligand secretion per unit area and DL is the diffusion coefficient of the ligand. The actual value of [L]S relative to the maximum is found to be a function of only two dimensionless variables: the ratio of receptor and ligand synthesis rates (equal to 1 for the plotted results) and a parameter that characterizes the efficiency of the receptor-mediated ligand capture (defined as ![]() where R0 = VR/kt; values of 100, 10, 1, and 0.1 were used here).
where R0 = VR/kt; values of 100, 10, 1, and 0.1 were used here).
Diffusion and mass transport
Biochemical rate processes depend on local species concentrations, which are not necessarily constant. When significant concentration gradients exist, it is appropriate to include a spatial dependence in the conservation equations, which then are solved in conjunction with appropriate boundary conditions that together specify the problem (Fig. 2b). For a species in solution present at a concentration Q(x, t), where x is a spatial position vector (coordinates in x, y, z) and t is time, the conservation equation is represented precisely as follows:
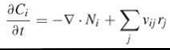
Here, Ni is a vector that specifies the outward flux of species i; thus, taking its gradient ![]() and adding a minus sign gives the net rate of species i into location x by mass transport. The second term accounts for the net generation of species i by reactions j, defined by the aforementioned rate laws rj and stoichiometry coefficients vij. Concentrations of species associated with membranes usually are expressed on a per unit surface area basis, and a planar geometry generally is adopted for the membrane.
and adding a minus sign gives the net rate of species i into location x by mass transport. The second term accounts for the net generation of species i by reactions j, defined by the aforementioned rate laws rj and stoichiometry coefficients vij. Concentrations of species associated with membranes usually are expressed on a per unit surface area basis, and a planar geometry generally is adopted for the membrane.
For molecules in solution, the flux Ni is the sum of two contributions: a convection term that accounts for the bulk flow of the fluid, given by vCi (v is the fluid velocity vector), and a diffusive flux term Ji that accounts for the tendency of molecules to disperse in solution. In the cellular context, it is important to note that the convective term also might include consideration of active transport processes, such as those actuated by motor proteins. To proceed, a semi-empirical constitutive equation that relates the form of Ji to macroscopic variables must be invoked. The only suitable theory that has been offered in that regard is attributed to Fick, who in 1855 asserted that the net diffusive flux of species i is proportional to its gradient in mole fraction; Einstein’s theory of Brownian motion, published 50 years later, is the microscopic analog of Fick’s Law. If species i is present in a dilute solution, Fick’s Law reduces to
![]()
The diffusion coefficient Di characterizes the mobility of species i in solution, determined by the thermal energy that promotes translational motion and the viscous drag force that opposes that motion. Substituting the expression above into the conservation equation and assuming that diffusion dominates over convection and Di is constant, one obtains the common reaction-diffusion equation:
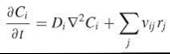
where ![]() is the Laplacian operator (sometimes written as ∆). Despite its lack of grounding in pure physical chemistry, which has been a source of some contention (16), Fick’s Law and the reaction-diffusion equation have been used with great success to characterize processes in the cell cytoplasm and at cell membranes. Given the empirical nature of Fick’s Law and the notable differences between molecular motions at microscopic (nm) versus macroscopic (μm) length scales, it is more appropriate to refer to Di as the apparent diffusivity or macroscopic mobility coefficient when applied to cell-associated molecules. An alternative approach, from the standpoint of modeling, is to develop a microscopic description of the system using Monte Carlo-based simulations (see the section “Mathematical Modeling Tools and Techniques” below).
is the Laplacian operator (sometimes written as ∆). Despite its lack of grounding in pure physical chemistry, which has been a source of some contention (16), Fick’s Law and the reaction-diffusion equation have been used with great success to characterize processes in the cell cytoplasm and at cell membranes. Given the empirical nature of Fick’s Law and the notable differences between molecular motions at microscopic (nm) versus macroscopic (μm) length scales, it is more appropriate to refer to Di as the apparent diffusivity or macroscopic mobility coefficient when applied to cell-associated molecules. An alternative approach, from the standpoint of modeling, is to develop a microscopic description of the system using Monte Carlo-based simulations (see the section “Mathematical Modeling Tools and Techniques” below).
Fluid mechanics and mechanical forces
Mechanical stress and strain have not been given due consideration in the broad context of cell biochemistry, yet mechanical forces can affect intracellular signaling processes in at least two distinct ways. First, at the level of macroscopic fluid flow, the conservation of fluid momentum determines the velocity field v, which along with active transport considerations determines the contribution of convective mass transport to the conservation of species i , as discussed in the previous section. Typically, this contribution to the reaction kinetics is neglected. Second, at the microscopic level, mechanical forces might alter directly the functions of macromolecules in prescribed ways, for example by exposing a cryptic binding pocket. This mode of regulation is thought to be at the core of mechanotransduction, which refers to the ability of cells to sense and respond to applied forces (17, 18).
Mathematical Modeling Tools and Techniques
Armed with reasonably good knowledge or hypotheses of the underlying biochemistry and the ability to formulate models based on physico-chemical principles, the models must be implemented to obtain and analyze the quantitative results and to generate predictions. Except in very simple, idealized cases, this task requires the use of various numerical methods and tools. In this section, an overview of the various model types and associated methods is offered.
Continuum models
The most common approach in mechanism-based modeling is to model the system as a continuum. The underlying assumption is that the state variables of the system (e.g., species concentrations) vary continuously in time and space and that, therefore, they evolve in a deterministic fashion, according to the aforementioned conservation equations and their initial and boundary constraints. The implementation of such models involves the solution of ordinary and partial differential equations (ODEs and PDEs, respectively). ODEs commonly are encountered in kinetic models of cell signaling systems in which the species concentrations are assumed to be spatially homogeneous within the cell/cell compartment or are averaged over the domain in an appropriate way. In kinetic models of even modest complexity, the ODEs are nonlinear and therefore must be solved using numerical methods. With the advent of efficient, implicit solution algorithms and steadily increasing computing power, handling even large systems of ODEs is straightforward and the integration of these algorithms in various software packages is widespread.
Solution of PDEs, commonly encountered in spatial modeling, is more complicated and computationally intensive by comparison, but several approximate numerical methods are available for this purpose. These include the finite difference, finite volume, and finite element methods, which vary according to their ease of implementation and applicability. The finite element method is the most complicated but generally applicable method for complex domain geometries; numerous software packages that implement this method are available, although the most powerful commercial packages are quite expensive. The freely available and user-friendly virtual cell software environment, which uses the finite volume method, has been used extensively for spatial modeling of intracellular processes (19). Several challenges are encountered in spatial modeling, regardless of the method used. First, the method and the accuracy of its computed results need to be validated. This requirement typically is achieved by testing the method on a simpler problem that has an analytical solution for comparison and by confirming that changes in the mesh size and time step do not affect the results significantly. Second, the amount of computation needed rises dramatically as the geometric complexity of the model increases from one up to three spatial coordinates, and a temptation exists to oversimplify the domain geometry to reduce the dimensionality of the problem. Finally, certain cell biological processes, such as cell motility, demand advanced modeling features such as the coupling of cell mechanics and chemical kinetics and the handling of moving boundaries, which can make the problem computationally prohibitive without the introduction of simplifying assumptions.
Stochastic modeling techniques
By comparison with continuum models, stochastic models aim to account for the inherent fluctuations and discrete nature of chemically reacting molecular systems. In cell biology, this approach has been applied most prominently in the arena of gene regulation (20-22) because these systems tend to be characterized by relatively small numbers of molecules (<100) and nonlinear, switch-like transitions. For stochastic systems that vary in time only, the most commonly used method for computing probabilistic realizations of such a model is the Monte Carlo-based algorithm developed by Gillespie (23), wherein running totals of the numbers of molecules in each state are tracked and adjusted in each time step according to probabilities of the various rate processes. This method is easy to implement and exact, but it is computationally inefficient for so-called stiff problems that possess a broad range of characteristic time scales. Approximate adaptations of the technique, using a method called tau leaping, allows more efficient computation of stiff systems albeit with a potential loss in accuracy (24, 25). Another approximate approach, valid in the limit of large numbers of molecules, is the stochastic differential equation method, which also is referred to as the chemical master equation or chemical Langevin equation approach. In this method, the probabilities of the various states being populated by different numbers of molecules evolve in time according to ODEs. Although this method is not necessarily more efficient or applicable to stiff systems, it serves as the starting point for several powerful approximations that are valid in certain limiting cases (20).
Methods for modeling spatially fluctuating reaction-diffusion systems, wherein molecules are treated as discrete particles whose coordinates are tracked and moved with time, also are available. These methods account for stochastic transitions as motivated above and are appropriate when concentration gradients are expected, as in the case of diffusion-controlled reactions. They also allow one to generate movies of the simulations to visualize the constituent processes in action. In the lattice Monte Carlo method, particles occupy nodes on a regularly spaced grid and move to adjacent nodes according to probabilities determined by the diffusion coefficient (and by long-range intermolecular potentials, if applicable). Changes in state occur probabilistically, and intermolecular interactions are subject to specific rules. For example, when the molecules are treated as point particles, interactions might occur in the next time step if the two species occupy adjacent nodes. In this limit, the method is very efficient computationally but suffers in accuracy because of its inability to resolve off-lattice events, which is especially significant when concentration gradients are steep. The Brownian dynamics or dissipative particle dynamics method is similar to the lattice Monte Carlo except that the particles adopt coordinates that are continuous throughout the domain. Thus, this method is accurate but also more computationally intensive. Both methods have been used in the context of signal transduction, particularly to study processes at cell membranes (26, 27).
Construction of biochemical networks
The multiplicity of modifications and interactions that signaling molecules engage in constitutes a large number of potential activity states. For instance, a protein or protein complex with 10 phosphorylation sites has 210 = 1,024 distinct combinations of modified or unmodified sites, and this number does not even include the binding status of each modified site. This issue, termed combinatorial complexity, presents a major challenge for detailed kinetic modeling, and hence rule-based modeling methods and accompanying software tools have been developed (28-30). In this approach, the individual interactions and reactions in the network are enumerated and the specific contexts in which they are allowed to occur (rules) are specified. The network of possible species, which could number in the hundreds or thousands, is generated automatically in the form of a system of ODEs; alternatively, the rules can be used in a stochastic simulation wherein the species of the network are formed spontaneously. Efforts are underway to extend this approach to the spatial domain as well.
Applications of Mathematical Modeling in Cell Signaling
With the availability of computational tools and, more importantly, the ability to judiciously apply physico-chemical principles to the biologic realm, what systems are and have been ripe for quantitative modeling? In this section, signal transduction processes that have been modeled successfully from the standpoint of yielding biologically meaningful insights are surveyed. Limitations on the number of references preclude a comprehensive coverage of this literature.
Models of specific signaling pathways and processes
For several reasons, no pathway has received more attention from modelers recently than the mitogen-activated protein kinase (MAPK) cascade, which involves the successive activation of three enzymes (Fig. 3). MAPKs are conserved in eukaryotes from yeast to man and are critical for cell functions that range from proliferation to differentiation to stress responses, among others. MAPKs are considered master integrators of upstream signals and master controllers of transcription factors and other downstream effectors. Aside from their obvious importance in cell biology, MAPK cascades have achieved “uber-pathway” status among modelers because of their potentially interesting dynamical properties. Building on pioneering work by Goldbeter and Koshland (31), who examined a hypothetical sequence of reversible enzymatic steps, the first model of a MAPK cascade was offered by Huang and Ferrell (32), who showed that the pathway was capable of a sensitive, switch-like relationship between the input (upstream of the cascade) and output (activation of MAPK); the basis for this response was the assumed biochemical mechanism, whereby MAPK and the upstream kinase are each activated by dual phosphorylation steps in a nonpro-cessive fashion. Building on this finding, more recent modeling efforts have focused on the consequences of the negative feedback regulation of the cascade (33-35) and the possibility that MAPK cascades operate as bistable switches, wherein two stable states (low and high MAPK activation) can be achieved at the same input strength (36, 37). Because of these modeling efforts, a solid understanding exists of what the cascade is capable of and how it apparently is modulated. Other models have been used to study the dynamics of MAPK cascades in the context of a full pathway, initiated by the activation of a specific receptor (38, 39). This approach is equally insightful albeit less general.
Another proving ground for mathematical modeling in cell signaling has been the dynamics of intracellular calcium. Spurred by the nonlinear nature of the calcium release mechanisms, which exhibit cooperativity and both positive and negative feedbacks, and by the availability of fluorescent dyes for imaging Ca2+ in living cells, both kinetic and spatial models of calcium dynamics have been explored in fine detail. Analyzed most extensively have been the mechanisms that give rise to a single calcium spike or sustained Ca2+ oscillations (40-44) and the propagation of calcium waves (45-49). These and other calcium models have proved useful in the fields of neuroscience, immunology, and cardiac biology.
Other receptor-proximal signal transduction interactions and pathways to which mathematical modeling and/or analysis have been applied successfully include the phosphoinositide 3-kinase pathway (50-54), which is central for signaling numerous responses including directed cell migration (chemotaxis), survival, and proliferation; the phospholipase C pathway/inositol cycle (44, 55, 56), which is important for intracellular calcium release and cell migration; and the actions and regulation of nonreceptor tyrosine kinases and protein phosphatases (57-60). Further downstream, models describing the regulation of transcription factors, most notably NFKB (61) and the STATs (signal transducing activators of transcription) (62), have been offered.

Figure 3. The MAPK cascade. A signaling cascade generally refers to a series of enzyme modification processes, as in the activation of extracellular signal-regulated kinase (Erk) isoforms, which are mammalian mitogen-activated protein kinases (MAPKs). The first kinase, Raf, is activated (indicated by an asterisk) by numerous inputs, allowing it to phosphorylate MEK on two sites; Erk is phosphorylated dually in a similar fashion by MEK. Each phosphorylation event is thought to require a separate encounter between enzyme and substrate, which gives rise to interesting dynamical properties. Not depicted here, but equally important, are the phosphatases that catalyze the reverse reactions.
Crosstalk in signal transduction networks
It has long been appreciated that signaling pathways/cascades do not operate in isolation. As considered theoretically by Bray (63), the use of multiple intermediates in signaling pathways affords more opportunities for regulation, often from parallel pathways (crosstalk). In fact, most pathways as classically defined are simply dominant routes of regulation embedded in larger networks of interactions in which proteins may interact with and/or modify multiple substrates (branch points) and receive regulatory inputs from multiple molecular partners (convergence points). Although experimental characterization of this important layer of complexity remains on the horizon, several groups have begun collecting data and building models of more complete signal transduction networks (38, 64, 65).
Pathway models related to specific cell responses
Signaling pathways drive phenotypic responses, but the precise details of how this occurs at the molecular level remain elusive. Consider cell migration. From the signaling literature, we know which receptor-mediated pathways are more or less important in controlling migration, and from the literature on cytoskeletal and adhesion dynamics we have a good understanding of how migration is coordinated, but currently the interface between those two fields is poorly understood by comparison. To move forward with a mathematical model capable of yielding mechanistic insights, the prudent course of action is to focus on the molecular details of the upstream signaling and use a coarse-grained, phenomenological model of the cell response or vice versa. In the context of cell migration and chemotaxis signaling, both approaches have been adopted in recent models (66-68). In other cell response-specific models, understandably more attention is paid to the execution of the response and less to the dynamics of the upstream regulation, as in the modeling of the cell cycle (69) and programmed cell death (70, 71). It is expected that such models will be refined as the details of how upstream signaling pathways regulate the execution of cell responses are revealed, and quantitative modeling and analysis could play a key role in elucidating those mechanisms.
Prospects and Challenges
Quantitative models of signal transduction processes, in conjunction with quantitative experimentation, are being used to evaluate biochemical mechanisms, predict the outcomes of novel experiments, and generate nonintuitive insights and hypotheses warranting additional study. One challenge we now face is how best to integrate such models to analyze complex intracellular and cell-cell communication systems. Although it is envisioned that quantitative models of cell physiology ultimately will progress in lock step with our knowledge of biochemical mechanisms, several hurdles loom on the horizon. Arguably the most important of these hurdles is the specification of parameter values and parameter estimation from data sets. With a few notable exceptions, mining the literature for “known” parameters is a dicey prospect, and this practice should be used only to provide reasonable, order-of-magnitude guesses for parameter values. Estimating parameters in their particular intracellular context would be ideal, and algorithms for doing so are applied readily, but a significant amount of quantitative data is needed. This exercise is only tractable for models with a modest number of parameters, and hence the challenge arises; a very detailed signaling network model might require hundreds of free parameters. At the pathway/network level, the formulation of simplified, coarse-grain models with lumped parameters offers a reasonable compromise. Even as experimental techniques for quantifying cellular processes become increasingly refined, the formulation of a useful model will continue to rely on biological and mathematical intuition, which, at its core, is the art of modeling.
References
1. Bray D. Reductionism for biochemists: how to survive the protein jungle. Trends Biochem. Sci. 1997; 22:325-326.
2. Sachs K, Gifford D, Jaakkola T, Sorger P, Lauffenburger DA. Bayesian network approach to cell signaling pathway modeling. Science STKE 2002; 148:pe 38.
3. Janes KA, Yaffe MB. Data-driven modelling of signal-transduction networks. Nat. Rev. Mol. Cell. Biol. 2006; 7:820-828.
4. Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2000; 103:211-225.
5. Mellman I. Endocytosis and molecular sorting. Annu. Rev. Cell Dev. Biol. 1996; 12:575-625.
6. Wiley HS, Shvartsman SY, Lauffenburger DA. Computational modeling of the EGF-receptor system: a paradigm for systems biology. Trends Cell Biol. 2003; 13:43-50.
7. Pawson T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 2004; 116:191-203.
8. Bhattacharyya RP, Remenyi A, Yeh BJ, Lim WA. Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu. Rev. Biochem. 2006; 75:655-680.
9. Sheetz MP. Glycoprotein motility and dynamic domains in fluid plasma membranes. Annu. Rev. Biophys. Biomol. Struct. 1993; 22:417-431.
10. Jacobson K, Dietrich C. Looking at lipid rafts? Trends Cell Biol. 1999; 9:87-91.
11. Hancock JF. Lipid rafts: contentious only from simplistic standpoints. Nat. Rev. Mol. Cell. Biol. 2006; 7:456-462.
12. Kusumi A, Nakada C, Ritchie K, Murase K, Suzuki K, Murakoshi H, Kasai RS, Kondo J, Fujiwara T. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: High-speed single-molecule tracking of membrane molecules. Annu. Rev. Biophys. Biomol. Struct. 2005; 34:351-U54.
13. Bevan AP, Drake PG, Bergeron JJM, Posner BI. Intracellular signal transduction: the role of endosomes. Trends Endocrin. Metabol. 1996; 7:13-21.
14. Haugh JM. Localization of receptor-mediated signal transduction pathways: the inside story. Mol. Interv. 2002; 2:292-307.
15. Bivona TG, Philips MR. Ras pathway signaling on endomembranes. Curr. Opin. Cell Biol. 2003; 15:136-142.
16. Agutter PS, Malone PC, Wheatley DN. Intracellular transport mechanisms: a critique of diffusion theory. J. Theor. Biol. 1995; 176:261-272.
17. Ingber DE. Cellular mechanotransduction: putting all the pieces together again. FASEB J. 2006; 20:811-827.
18. Orr AW, Helmke BP, Blackman BR, Schwartz MA. Mechanisms of mechanotransduction. Dev. Cell 2006; 10:11-20.
19. Slepchenko BM, Schaff JC, Macara I, Loew LM. Quantitative cell biology with the Virtual Cell. Trends Cell Biol. 2003; 13:570-576.
20. Kepler TB, Elston TC. Stochasticity in transcriptional regulation: Origins, consequences, and mathematical representations. Bio- phys. J. 2001; 81:3116-3136.
21. Rao CV, Wolf DM, Arkin AP. Control, exploitation and tolerance of intracellular noise. Nature 2002; 420:231-237.
22. Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: From theories to phenotypes. Nat. Rev. Genetics 2005; 6:451-464.
23. Gillespie DT. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977; 81:2340-2361.
24. Gillespie DT. Approximate accelerated stochastic simulation of chemicallyreacting systems. J. Chem. Phys. 2001; 115:1716-1733.
25. Rathinam M, Petzold LR, Cao Y, Gillespie DT. Stiffness in stochastic chemically reacting systems: The implicit tau-leaping method. J. Chem. Phys. 2003; 119:12784-12794.
26. Shea LD, Omann GM, Linderman JJ. Calculation of diffusion-limited kinetics for the reactions in collision coupling and receptor cross-linking. Biophys. J. 1997; 73:2949-2959.
27. Monine MI, Haugh JM. Reactions on cell membranes: Comparison of continuum theory and Brownian dynamics simulations. J. Chem. Phys. 2005; 123:074908.
28. Hlavacek WS, Faeder JR, Blinov ML, Posner RG, Hucka M, Fontana W. Rules for modeling signal-transduction systems. Sci. STKE 2006; 344:re6.
29. Lok L, Brent R. Automatic generation of cellular reaction networks with Moleculizer 1.0. Nat. Biotechnol. 2005; 23:131-136.
30. Conzelmann H, Saez-Rodriguez J, Sauter T, Kholodenko BN, Gilles ED. A domain-oriented approach to the reduction of combinatorial complexity in signal transduction networks. BMC Bioinformatics 2006; 7:34.
31. Goldbeter A, Koshland DE. Jr An amplified sensitivity arising from covalent modification in biological systems. Proc. Natl. Acad. Sci. U.S.A. 1981; 78:6840-6844.
32. Huang CF, Ferrell JE Jr. Ultrasensitivity in the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:16468-10083.
33. Kholodenko BN. Negative feedback and ultrasensitivity can bring about oscillations in the mitogen-activated protein kinase cascades. Eur. J. Biochem. 2000; 267:1583-1588.
34. Asthagiri AR, Lauffenburger DA. A computational study of feedback effects on signal dynamics in a mitogen-activated protein kinase (MAPK) pathway model. Biotechnol. Prog. 2001; 17:227- 239.
35. Behar M, Hao N, Dohlman HG, Elston TC. Mathematical and computational analysis of adaptation via feedback inhibition in signal transduction pathways. Biophys. J. 2646; 93:806-821.
36. Markevich NI, Hoek JB, Kholodenko BN. Signaling switches and bistability arising from multisite phosphorylation in protein kinase cascades. J. Cell Biol. 2004; 164:353-359.
37. Wang X, Hao N, Dohlman HG, Elston TC. Bistability, stochasticity, and oscillations in the mitogen-activated protein kinase cascade. Biophys. J. 2006; 90:1961-1978.
38. Bhalla US, Ram PT, Iyengar R. MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science 2002; 297:1018-1023.
39. Schoeberl B, Eichler-Jonsson C, Gilles ED, Muller G. Computational modeling of the dynamics of the MAP kinase cascade activated by surface and internalized EGF receptors. Nat. Biotech- nol. 2002; 20:370-375.
40. Meyer T, Stryer L. Molecular model for receptor-stimulated calcium spiking. Proc. Natl. Acad. Sci. U.S.A. 1988; 85:5051-5055.
41. Goldbeter A, Dupont G, Berridge MJ. Minimal model for signal-induced Ca2+ oscillations and for their frequency encoding through protein phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 1990; 87:1461-1465.
42. Tang YH, Stephenson JL, Othmer HG. Simplification and analysis of models of calcium dynamics based on IP3-sensitive calcium channel kinetics. Biophys. J. 1996; 70:246-263.
43. Schuster S, Marhl M, Hofer T. Modelling of simple and complex calcium oscillations - From single-cell responses to intercellular signalling. Eur. J. Biochem. 2002; 269:1333-1355.
44. Xu C, Watras J, Loew LM. Kinetic analysis of receptor-activated phosphoinositide turnover. J. Cell Biol. 2003; 161:779-791.
45. Sneyd J, Keizer J, Sanderson MJ. Mechanisms of calcium oscillations and waves: a quantitative analysis. FASEB J. 1995; 9:1463- 1472.
46. Falcke M, Tsimring L, Levine H. Stochastic spreading of intracellular Ca2+ release. Phys. Rev. E 2000; 62:2636-2643.
47. Sobie EA, Dilly KW, Cruz JD, Lederer WJ, Jafri MS. Termination of cardiac Ca2+ sparks: An investigative mathematical model of calcium-induced calcium release. Biophys. J. 2002; 83:59-78.
48. Fink CC, Slepchenko B, Moraru II, Watras J, Schaff JC, Loew LM. An image-based model of calcium waves in differentiated neuroblastoma cells. Biophys. J. 2000; 79:163-183.
49. Koh XY, Srinivasan B, Ching HS, Levchenko A. A 3D Monte Carlo analysis of the role of dyadic space geometry in spark generation. Biophys. J. 2006; 90:1999-2014.
50. Park CS, Schneider IC, Haugh JM. Kinetic analysis of platelet-derived growth factor receptor/phosphoinositide 3-kinase/Akt signaling in fibroblasts. J. Biol. Chem. 2003; 278:37064-37072.
51. Ma L, Janetopoulos C, Yang L, Devreotes PN, Iglesias PA. Two complementary, local excitation, global inhibition mechanisms acting in parallel can explain the chemoattractant-induced regulation of PI(3,4,5)P3 response in Dictyostelium cells. Biophys. J. 2004; 87:3764-3774.
52. Skupsky R, Losert W, Nossal RJ. Distinguishing modes of eukaryotic gradient sensing. Biophys. J. 2005; 89:2806-2823.
53. Schneider IC, Haugh JM. Quantitative elucidation of a distinct spatial gradient-sensing mechanism in fibroblasts. J. Cell Biol. 2005; 171:883-892.
54. Kaur H, Park CS, Lewis JM, Haugh JM. Quantitative model of Ras/phosphoinositide 3-kinase signalling cross-talk based on co-operative molecular assembly. Biochem. J. 2006; 393:235-243.
55. Haugh JM, Wells A, Lauffenburger DA. Mathematical modeling of epidermal growth factor receptor signaling through the phospholipase C pathway: mechanistic insights and predictions for molecular interventions. Biotechnol. Bioeng. 2000; 70:225-238.
56. Narang A, Subramanian KK, Lauffenburger DA. A mathematical model for chemoattractant gradient sensing based on receptor- regulated membrane phospholipid signaling dynamics. Ann. Biomed. Eng. 2001; 29:677-691.
57. Wofsy C, Vonakis BM, Metzger H, Goldstein B. One Lyn molecule is sufficient to initiate phosphorylation of aggregated high-affinity IgE receptors. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:8615-8620.
58. Heinrich R, Neel BG, Rapoport TA. Mathematical models of protein kinase signal transduction. Mol. Cell 2002; 9:957-970.
59. Faeder JR, Hlavacek WS, Reischl I, Blinov ML, Metzger H, Redondo A, Wofsy C, Goldstein B. Investigation of early events in FceRI-mediated signaling using a detailed mathematical model. J. Immunol. 2003; 170:3769-3781.
60. Barua D, Faeder JR, Haugh JM. Structure-based kinetic models of modular signaling protein function: focus on Shp2. Biophys. J. 2646; 92:2290-2300.
61. Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkB-NF- kB signaling module: temporal control and selective gene activation. Science 2002; 298:1241-1245.
62. Yamada S, Shiono S, Joo A, Yoshimura A. Control mechanism of JAK/STAT signal transduction pathway. FEBS Lett. 2003; 534:190-196.
63. Bray D. Intracellular signaling as a parallel distributed process. J. Theor. Biol. 1990; 143:215-231.
64. Hatakeyama M, Kimura S, Naka T, Kawasaki T, Yumoto N, Ichikawa M, Kim J, Saito K, Saeki M, Shirouzu M, et al. A computational model on the modulation of mitogen-activated protein kinase (MAPK) and Akt pathways in heregulin-induced ErbB signalling. Biochem. J. 2003; 373:451-463.
65. Kiyatkin A, Aksamitiene E, Markevich NI, Borisov NM, Hoek JB, Kholodenko BN. Scaffolding protein Grb2-associated binder 1 sustains epidermal growth factor-induced mitogenic and survival signaling by multiple positive feedback loops. J. Biol. Chem. 2006; 281:19925-19938.
66. Haugh JM. Deterministic model of dermal wound invasion incorporating receptor-mediated signal transduction and spatial gradient sensing. Biophys. J. 2006; 90:2297-2308.
67. Maree AFM, Jilkine A, Dawes A, Grieneisen VA, Edelstein-Keshet L. Polarization and movement of keratocytes: A multiscale modelling approach. Bull. Math. Biol. 2006; 68:1169-1211.
68. Dawes AT, Edelstein-Keshet L. Phosphoinositides and Rho proteins spatially regulate actin polymerization to initiate and maintain directed movement in a one-dimensional model of a motile cell. Biophys. J. 2646; 92:744-768.
69. Csikasz-Nagy A, Battogtokh D, Chen KC, Novak B, Tyson JJ. Analysis of a generic model of eukaryotic cell-cycle regulation. Biophys. J. 2006; 90:4361-4379.
70. Fussenegger M, Bailey JE, Varner J. A mathematical model of caspase function in apoptosis. Nature Biotechnol. 2000; 18:768-774.
71. Eissing T, Conzelmann H, Gilles ED, Allgower F, Bullinger E, Scheurich P. Bistability analyses of a caspase activation model for receptor-induced apoptosis. J. Biol. Chem. 2004; 279:36892-36897.
Further Reading
Kholodenko BN. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell. Biol. 2006; 7:165-176.
Lauffenburger DA, Linderman JJ. Receptors: Models for Binding, Trafficking, and Signaling. 1993. Oxford University Press, New York.
Ma’ayan A, Blitzer R.D., Iyengar R. Toward predictive models of mammalian cells. Annu. Rev. Biophys. Biomol. Struct. 2005; 34:319-349.
Mogilner A, Wollman R, Marshall WF. Quantitative modeling in cell biology: what is it good for? Dev. Cell 2006; 11:279-287.
Slepchenko BM, Schaff JC, Carson JH, Loew LM. Computational cell biology: spatiotemporal simulation of cellular events. Annu. Rev. Biophys. Biomol. Struct. 2002; 31:423-441.
Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell Biol. 2003; 15:221-231.
See Also
Cell Cycle, Computation and Modeling of
Chemotaxis
Receptor-Ligand Interactions
Signal Cascades, Protein Interaction Networks in
Systems Biology