Microreactors in Organic Chemistry and Catalysis, Second Edition (2013)
7. Heterogeneous Reactions
7.9. Multistep Synthesis
The multistep synthesis of structurally complex molecules from readily available substrates is one of the ideal methods and ongoing challenges of synthetic chemistry. The continuous flow systems are considerably well suited to perform multistep synthesis [6, 15, 175, 176]. Reagents can be introduced into the stream of reactants anywhere in the flow system at precisely the time that is required for the reaction. For multistep continuous flow syntheses, all the substrates, reagents, catalysts, and solvents used in the first step must be compatible with all the subsequent reaction steps. Alternatively, appropriate scavengers or microextractors must be set up. In addition, if an in-series connection of more than two flow reactors is arranged, the flow rates and back pressures also have to be adjusted accordingly. By employing several flow reactors, scavengers, and/or microextractors in a linear arrangement, space integration of the reactions can be designed.
Kappe et al. described a two-step continuous-flow synthesis of 2-amino-4′-chlorobiphenyl, which is a key intermediate for the industrial synthesis of fungicide Boscalid (Scheme 7.46) [177]. Homogeneous palladium-catalyzed Suzuki–Miyaura coupling was accomplished by using an X-Cube reactor. The crude coupling adduct was then reduced by using an H-Cube equipped with a Pt/C cartridge as a heterogeneous catalyst. They made clear that contamination of the homogeneous Pd metal for the cross-coupling into the Pt/C catalyst cartridge resulted in poor productivity in the second reducing step. To eliminate the palladium residue, a Pd-scavenger cartridge, Quadrapure-TU, was placed in-line between the X-Cube and H-Cube reactors.
Scheme 7.46 Two-step continuous-flow synthesis of Boscalid intermediate.
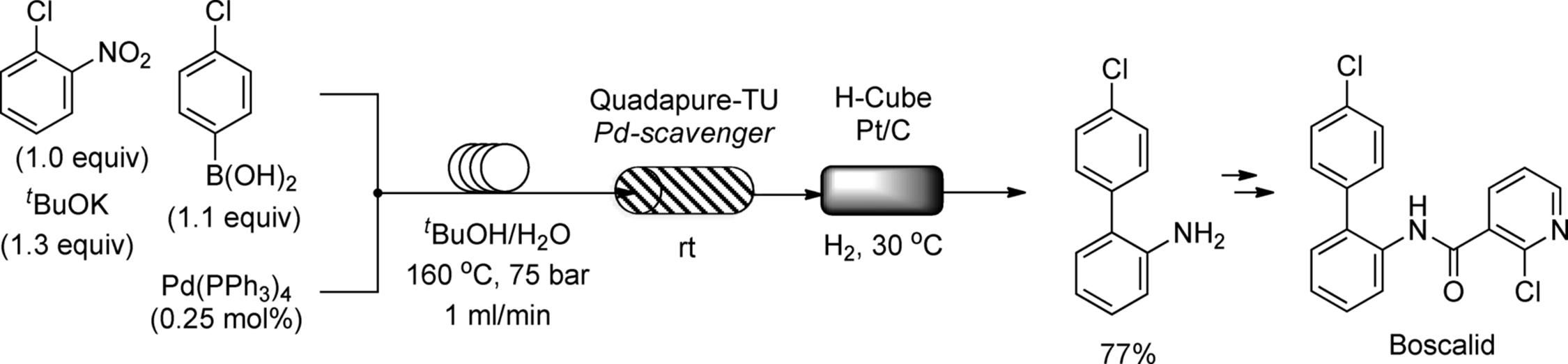
Ulven's group synthesized a library of heterocyclic compounds that were potential candidates for chemokine receptor CCR8 ligands by using a multistep continuous-flow system [178]. The flow system consisted of one homogeneous T-piece reactor, two heterogeneous reactors, and two scavengers, in which urea formation, Cbz-deprotection, and N-benzylation were performed, and is shown in Scheme 7.47. After the reaction of Cbz-protected diamine (66) with isocyanate (67) occurred at 75 °C for 7 min (residence time) in the first reactor, unreacted isocyanate was scavenged in a column with trisamine on macroporous polystyrene (MP-trisamine). The reaction stream was directed into an H-Cube reactor with a Pd/C cartridge that performed reductive deprotection of the Cbz group. Alkylation of the resulting compound (68) with benzylbromide (69) in the third reactor filled with polymer-supported base, followed by a second scavenging column to remove excess benzylbromide, afforded the desired heterocycles (70).
Scheme 7.47 Three-step continuous flow synthesis of drug candidates.
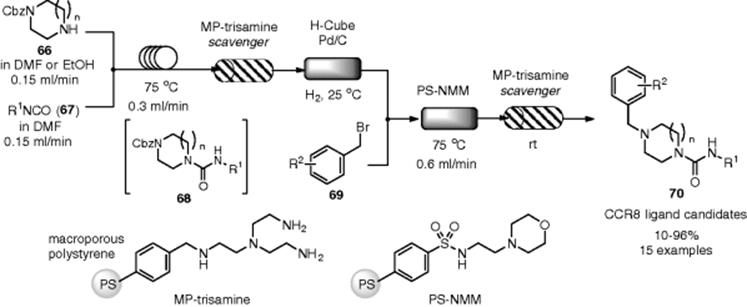
Perhaps, the most successful and elaborate example of a multistep flow synthesis was described by Ley and coworkers (Scheme 7.48). They achieved a continuous flow synthesis of the complex natural product oxamaritidine in excellent yield (>40%) without any intermediate isolation and without a traditional work-up at the intermediate stages [110]. Seven flow chemical transformations were assembled into a single flow system, which included various packed columns containing immobilized reagents, catalysts, scavengers, or catch and release agents. Azidation of benzylbromide (71) with polymer-supported azide to give the corresponding azide (72) initiated the flow sequence. Staudinger phosphinimine formation was achieved in the catch and release reactor followed by an aza-Wittig reaction with aldehyde (74), which was prepared separately by oxidation of (73). In the catch and release reactor, azide (72) was trapped by the immobilized phosphine reagent and then released by the reaction with aldehyde (74) to give imine (75). Imine (75) was reduced by an H-Cube hydrogenator to afford amine (76). Since the next reactions were incompatible with THF as a solvent, the solvent had to be changed to dichloromethane. N-protection of (76) was successively achieved through the homogeneous reactor. Then, phenolic oxidation of (77) by phenyliodine bis(trifluoroacetate) (PIFA) immobilized on polystyrene was performed under flow conditions. Successive N-deprotection of (78) facilitated the spontaneous 1,4-addition, which afforded the desired (±)-oxomaritidine (79) in 90% purity. The overall yield was calculated to be more than 40%. It is noteworthy that the entire sequence was completed in 6 h, while the total synthesis using the conventional batch reactions required approximately 4 days.
Scheme 7.48 Total synthesis of (±)-oxomaritidine under flow conditions.
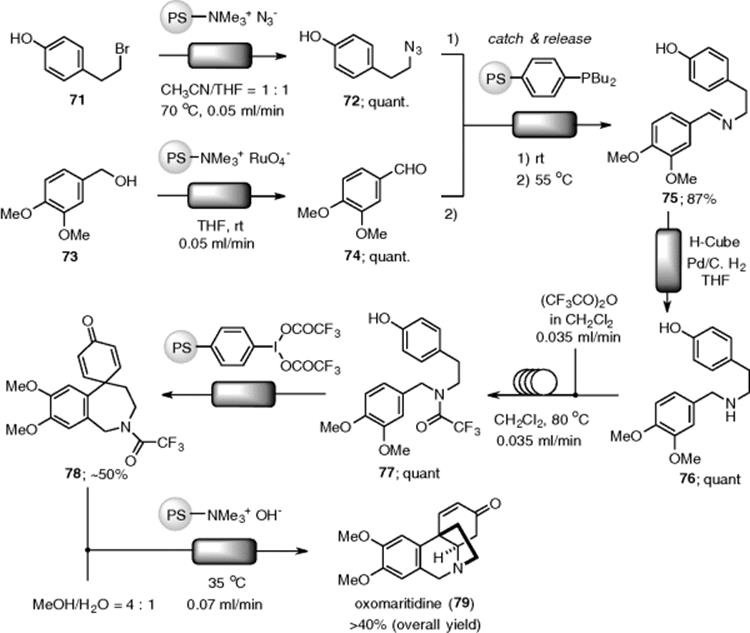
A multistep flow synthesis of the tyrosine kinase inhibitor Imatiniv (87) was developed by the same group (Scheme 7.49) [179]. Imatiniv, marketed as Gleevec, is a targeted cancer drug used for the treatment of several different cancers. The first step, amidation of aniline (81) with acid chloride (80) in CH2Cl2, was accomplished by the catch and release protocol through a polymer-supported DMAP packed-bed reactor. The excess acid chloride was in-line removed through a scavenger column (QP-DMA). Amide (82) could be directly isolated following solvent evaporation in 78% yield and excellent purity (>95%). The reaction solvent in the second step, the SN2 reaction of (82) with amine (83), had to be changed to a DMF/CH2Cl2 mixture due to the poor solubility of (82) in CH2Cl2. Although they planned to combine the secondary stream of (83) in DMF directly with that of (82) in CH2Cl2, a control of the stoichiometry of the two flows was not possible. Therefore, the authors employed an autosampler attached to a UV spectrometer that automatically submitted fractions of different concentrations to the next step. Nitrogen gas was bubbled into the solution to remove CH2Cl2 as it was collected to perform the solvent exchange. The resulting solution of a mixture of (82) and (83) was directed into a CaCO3-fixed column followed by two columns to afford (84) in 80% yield with >95% purity. The isocyanate-based column acted as a scavenger to remove unreacted piperazine. Sulfonic acid resin was used as a catch and release purification column, which trapped intermediate (84) with any unreacted (82) simply passing through to waste. Buchwald–Hartwig coupling of (84) with (86) was employed as the final step to produce (87). Unfortunately, the same conditions in the batch mode could not be applied under flow conditions due to solubility problems and precipitation issues. Finally, they found that a 1,4-dioxane/tBuOH solvent system was ideal for dissolving all the substrates and products and could be heated to the high temperatures required for the reaction. Multistep synthesis was completed by Buchwald–Hartwig coupling using BrettPhos-Pd precatalyst (85) under homogeneous flow conditions. After the flow sequence, automatic column chromatography afforded the desired compound (87) in 32% overall yield.
Scheme 7.49 Flow process for the synthesis of Gleevec 87.
Recently, many examples of multistep flow synthesis have been extensively described. For additional examples of multistep flow syntheses, the reader is kindly referred to the additional references [180–190].