Medical Microbiology
Section 5 Diagnosis and control
31 Diagnosis of infection and assessment of host defence mechanisms
Introduction
Good quality specimens are needed for reliable microbiologic diagnoses
The precise identification of the causative organism in infection has become increasingly important now that therapeutic intervention is possible. The ability to achieve this depends upon a positive interaction between the clinician and the microbiologist; the clinician must be aware of the complexity of the tests and the time required to achieve a result. In turn, the microbiologist must appreciate the nature of the patient’s condition and be able to assist the clinician in interpreting the laboratory report. A fundamental step in any diagnosis is the choice of an appropriate specimen, which ultimately depends upon an understanding of the pathogenesis of infections.
Microbiology differs from other clinical laboratory disciplines in the amount of interpretative input required. When a specimen is received, decisions are made regarding the appropriate processing pathway, and when the result is received, it must be interpreted in relation to the specimen and the patient.
Aims of the clinical microbiology laboratory
The aims of the microbiology laboratory are:
• to provide accurate information about the presence or absence of microorganisms in a specimen that may be involved in a patient’s disease process
• where relevant, to provide information on the antimicrobial susceptibility of the microorganisms isolated.
Identification is achieved by detecting the microorganism or its products or the patient’s immune response
Laboratory tests are carried out:
• to detect microorganisms or their products in specimens collected from the patient
• to detect evidence of the patient’s immune response (production of antibodies) to infection.
While there are different protocols for different specimens (e.g., urine, faeces, genital tract, blood, etc.), the tests fall into three main categories:
1. Identification of microorganisms by isolation and culture. Microorganisms may grow in artificial media or, in the case of viruses, in cell cultures. In some instances, quantification is important (e.g. more than 105 bacteria/mL of urine is indicative of infection whereas lower numbers are not; see Ch. 20). Once an organism has been isolated in culture, its susceptibility to antimicrobial agents can be determined.
2. Identification of a specific microbial gene or product. Non-cultural techniques that do not depend upon the growth and multiplication of microorganisms to detect microorganisms have the potential to yield more rapid results. These techniques include the detection of structural components of the cell (e.g. cell wall antigens) and extracellular products (e.g. toxins). Alternatively, molecular approaches are increasingly available such as the detection of specific gene sequences in clinical specimens using DNA probes or the polymerase chain reaction (PCR; see below). They are potentially applicable to all microorganisms, but antimicrobial susceptibilities cannot be determined without culture (although the presence of resistance genes may be detectable by specific probes).
3. Detection of specific antibodies to a pathogen. This is especially important when the pathogen cannot be cultivated in laboratory media (e.g. Treponema pallidum, many viruses) or when culture would be particularly hazardous to laboratory staff (e.g. culture of Francisella tularensis, the cause of tularaemia, or the fungus Coccidioides immitis). Detection of IgM and/or IgG antibodies in a single serum collected during the acute phase of illness can be helpful in diagnosis of, for example, rubella by specific IgM, hepatitis A by IgM and hepatitis B by HepB surface antigen, or in rare diseases such as Lassa fever. The classic diagnostic method is by detection of a rise (fourfold or greater) in antibody titre between ‘paired’ sera, collected in the acute phase of an infection (5–7 days after onset of symptoms) and in convalescence (after 3–4 weeks). Such tests therefore tend to result in a delayed or retrospective diagnosis and are therefore of limited help for clinical management.
Specimen processing
Specimen handling and interpretation of results is based upon a knowledge of normal flora and contaminants
Specimens intended for cultivation of microorganisms can be divided into two types:
• those from sites that are normally sterile
• those from sites that usually have a commensal flora (Box 31.1; see also Ch. 8).
A thorough knowledge of the microorganisms normally isolated from specimens from non-sterile sites, and the common contaminants of specimens collected from sterile sites, is important to ensure that specimens are properly handled and the results are correctly interpreted. Some specimens from sites that should be sterile (e.g. bladder urine, sputum from the lower respiratory tract) are usually collected after passage through orifices that have a normal flora, which may contaminate the specimens. This needs to be considered when interpreting the culture results of these specimens.
![]()
Box 31.1  Sampling Sites, The Normal Flora and Interpretation of Results
Sampling Sites, The Normal Flora and Interpretation of Results
Body sites that are normally sterile
• Blood and bone marrow
• Cerebrospinal fluid
• Serous fluids
• Tissues
• Lower respiratory tract
• Bladder
Body sites that have a normal commensal flora
• Mouth, nose and upper respiratory tract
• Skin
• Gastrointestinal tract
• Female genital tract
• Urethra
Some sites in the body are sterile in health so that growth of any organism is indicative of infection provided that the specimen has been properly collected and transported, and examined in the laboratory without delay. The significance of isolates from sites that have a commensal flora depends upon the identity of the isolate and the quantity, as well as the immune status of the patient.
![]()
In an ideal world, each specimen arriving in the laboratory would be considered in turn together with the information provided about the patient on the request form so that the microbiologist could assess the pathogens likely to be present and devise an ‘individualized’ processing plan. However, in reality, this approach is not practicable because of constraints on time and money. Thus, specimens tend to be processed by type (e.g. urine, blood, faeces) and the microbiologist looks for easily cultivated pathogens known to be associated with each sample type. However, if the laboratory is provided with suitable information, such as a statement of possible aetiology, more fastidious or unusual pathogens can be sought and relevant antibiotic susceptibilities assessed. To obtain a test result that correctly identifies the infection, it is important to collect an appropriate specimen, to use the appropriate transport conditions and to deliver specimens rapidly to the laboratory. These conditions all affect the accuracy of the laboratory report, and therefore its value to the clinician and ultimately to the patient. Key points to remember about specimen collection are summarized in Box 31.2.
![]()
Box 31.2 Important Steps in Specimen Collection and Delivery to The Laboratory
• Take the appropriate specimen, e.g. blood and cerebrospinal fluid in suspected meningitis.
• Collect the specimen at the appropriate time, during the acute phase of the disease, e.g. malarial films, virus isolation, viral genome detection, IgM detection.
• If possible, collect specimen before patient receives antimicrobials.
• Collect enough material and an adequate number of samples, e.g. enough blood/serum for more than one set of blood cultures.
• Avoid contamination:
• from normal flora, e.g. midstream urine
• from non-sterile equipment.
• Use the correct containers and appropriate transport media.
• Label specimens properly.
• Complete request form with enough clinical information and a statement of possible aetiology.
• Inform the laboratory if special tests are required.
• Transport specimens rapidly to the laboratory.
The responsibility of the clinician does not end with collection of the specimen and requesting tests. Good communication with the microbiologist is essential.
![]()
Routine culture takes at least 18 h to produce a result
Time is a key factor because the conventional methods of microbiologic diagnosis depend upon growth and identification of the pathogen. Results of routine culture cannot be achieved in < 18 h and may take much longer (e.g. several weeks) for a minority of pathogens such as the mycobacteria, which grow very slowly. Thus, specimen processing can be categorized according to the time required to achieve a result and the method – cultural or non-cultural. An alternative route to the diagnosis of an infection is an immunologic one, relying on the detection of an antibody response to the putative pathogen in the patient’s blood. These diagnostic routes are summarized in Figure 31.1, but rapid technologies (e.g. PCR, nucleic-acid probes, microarrays, etc.) have had a major influence on this process.
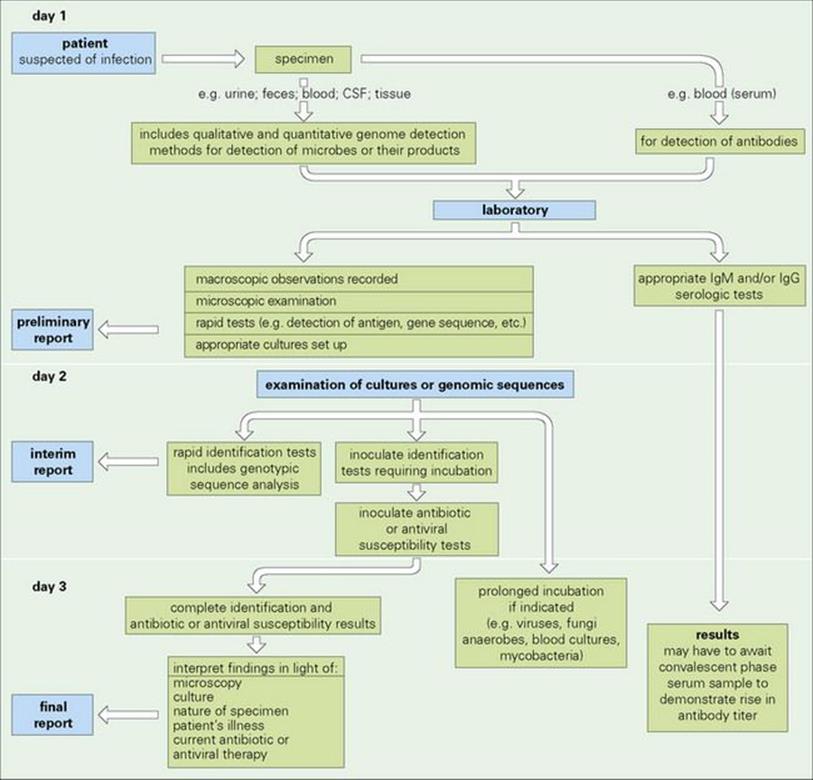
Figure 31.1 Route from patient to microbiologic diagnosis. This scheme shows a general overview of key steps in specimen processing. Some tests can be performed on the specimen immediately and yield ‘same day’ results. Culture of specimens usually involves a minimum of 18 h incubation before colonies are visible and can be identified. Antibiotic susceptibility tests involve a further incubation period. Alternatively, the diagnosis may be based on the detection of specific antibodies in serum samples: cerebrospinal fluid (CSF), genomic sequences, etc.
Non-cultural techniques for the laboratory diagnosis of infection
Non-cultural techniques do not require microorganism multiplication before detection
Although medical microbiology has long been synonymous with the cultivation of microorganisms from patients’ specimens, these techniques are labour-intensive and slow to produce results (days rather than hours) because replication of organisms is a necessary, but rate-limiting, step. In addition, some microorganisms cannot be cultured in artificial media, and viable organisms may be difficult to recover from specimens of patients who have received antimicrobial therapy. Non-cultural techniques do not require multiplication of the microorganism before its detection. Some techniques, such as microscopy and detection of microbial antigens in specimens, can provide very rapid results (i.e. within 2 h). Other non-cultural methods such as the use of DNA probes and amplification of DNA by the polymerase chain reaction (PCR) may also provide a rapid answer in a matter of hours.
Microscopy
Microscopy is an important first step in the examination of specimens
Microscopy plays a fundamental role in microbiology. Although microorganisms show a wide range in size (see Ch. 1) they are too small to be seen individually by the naked eye, and therefore a microscope is an essential tool in microbiology. The various types of microscopy are summarized in Figure 31.2. The light microscope magnifies objects and therefore improves the resolving power of the naked eye from about 100 000 nm (0.1 mm) to 200 nm; the electron microscope can improve this to 0.1 to 1.0 nm.
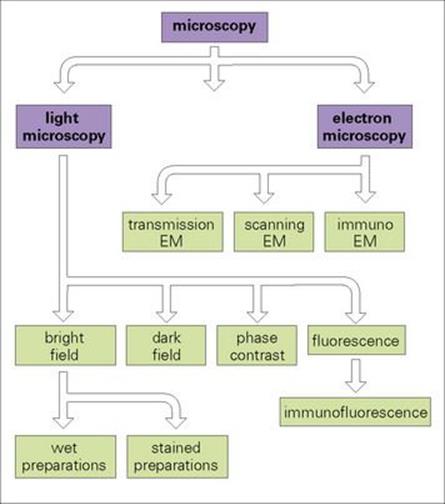
Figure 31.2 Applications of microscopy to microbiology. The scheme shows the different uses of light and electron microscopy (EM) for looking at microbes, although the latter is much less frequently used.
Light microscopy
Bright field microscopy is used to examine specimens and cultures as wet or stained preparations
Wet preparations are used to demonstrate:
• blood cells and microbes in fluid specimens such as urine, faeces or cerebrospinal fluid (CSF)
• cysts, eggs and parasites in faeces
• fungi in skin
• protozoa in blood and tissues.
Living organisms can be examined to detect motility.
Dyes are used to stain cells so that they can be seen more easily. Stains are usually applied to dried material that has been fixed (by heat or alcohol) onto the microscope slide. Samples from specimens themselves, or pure cultures, can be stained. The slide can then be viewed in the light microscope with an oil immersion lens, which improves the resolving power of the microscope.
The most important differential staining technique in bacteriology is the ‘Gram’ stain
Differential staining procedures exploit the fact that cells with different properties stain differently and thus can be distinguished. Based on their reaction to Gram’s stain (Fig. 31.3), bacteria are divided into two broad groups:
• Gram positive (stain purple)
• Gram negative (stain pink).
This difference is related to differences in the structure of the cell walls of the two groups (see Ch. 2).
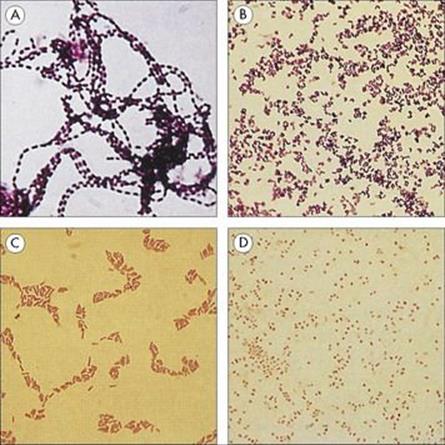
Figure 31.3 The Gram stain is the most important stain for studying bacteria. The combination of the violet dye (crystal violet) and iodine (acting as a mordant) binds to the cell wall. Gram-positive cells retain the stain when challenged with acetone and remain purple. Gram-negative cells lose the purple stain and appear colourless until stained with a pink counterstain (neutral red or safranin). Examination of Gram-stained films also allows the shape of the cells to be noted. Some examples are shown: (A) Gram-positive cocci in chains (streptococci); (B) Gram-positive rods (Listeria); (C) Gram-negative rods (E. coli); (D) Gram-negative cocci (Neisseria).
Acid-fast stains are used to detect mycobacteria
Some organisms, particularly mycobacteria, which have waxy cell walls, do not readily take up the Gram stain. To demonstrate their presence, special staining techniques are used which rely on the ability of such organisms to retain the stain in the presence of ‘decolourizing’ agents such as acid and alcohol. The Ziehl–Neelsen stain (see Fig. 19.20) is a classic differential staining procedure that uses heat to drive the fuchsin stain into the cells; mycobacteria stained with fuchsin withstand decolourization with acid and alcohol and are therefore known as ‘acid-’ and ‘alcohol-fast’, whereas other bacteria lose the stain after acid and alcohol treatment. Alternatively, many laboratories use the fluorescent dye auramine, which has a strong affinity for the waxy cell wall of mycobacteria, to demonstrate these organisms by fluorescence microscopy (Fig. 31.4).
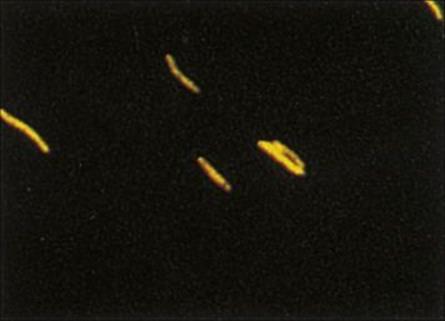
Figure 31.4 Fluorochrome stain of Mycobacterium tuberculosis with a mixture of auramine O and rhodamine B. Mycobacteria appear fluorescent under ultraviolet light.
(Courtesy of D.K. Banerjee.)
Other staining techniques can be used to demonstrate particular features of cells
Examples of such features to aid identification include the volutin (polyphosphate) storage granules in Corynebacterium spp. and lipid in Bacillus spp. (Fig. 31.5).
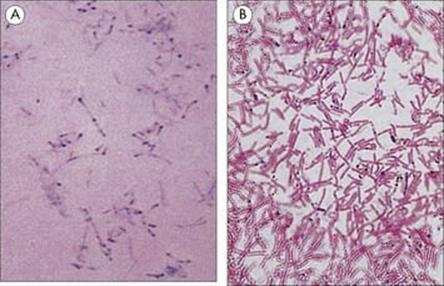
Figure 31.5 Special staining techniques can be used to demonstrate particular features of bacterial cells. (A) Corynebacteria stained to demonstrate polymetaphosphate storage granules (volutin granules), which appear as dark spots in blue-green cells (Albert’s stain). (B) Lipid storage granules in Bacillus cereus stained with Sudan black (black lipid against red cells).
Dark field (dark ground) microscopy is useful for observing motility and thin cells such as spirochetes
The light microscope may be adapted by modifying the condenser so that the object appears brightly lit against a dark background. Living organisms can be examined by dark field microscopy and thus motility can be observed. The method is also used for visualizing very thin cells such as spirochetes because the light reflected from the surface of the cells makes them appear larger and therefore more easily visible than when examined by bright field microscopy (Fig. 31.6).
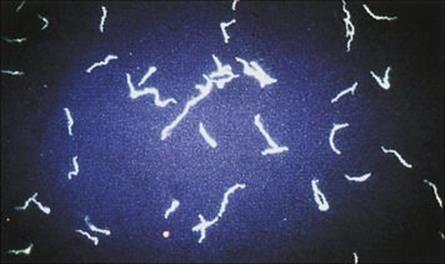
Figure 31.6 Spirochetes visualized by dark ground microscopy. Spirochetes and leptospires are much thinner than most bacterial cells (approximately 0.1 μm in diameter compared with 1 μm for E. coli), but they appear larger when viewed by dark ground illumination.
Phase contrast microscopy increases the contrast of an image
This technique enhances the very small differences in refractive index and density between living cells and the fluid in which they are suspended and therefore produces an image with a higher degree of contrast than that achieved by bright field microscopy.
Fluorescence microscopy is used for substances that are either naturally fluorescent or have been stained with fluorescent dyes
If light of one wavelength shines on a fluorescent object, it emits light of a different wavelength. Some biological substances are naturally fluorescent; others can be stained with fluorescent dyes and viewed in a microscope with an ultraviolet light source instead of white light (see Fig. 31.4).
Fluorescence microscopy is widely used in microbiology and immunology and has been developed to detect microbial antigens in specimens and tissues by ‘staining’ with specific antibodies tagged with fluorescent dyes (immunofluorescence). The method can be made more sensitive or can be adapted to the detection of antibody by labelling a second antibody in an indirect test (Fig. 31.7).
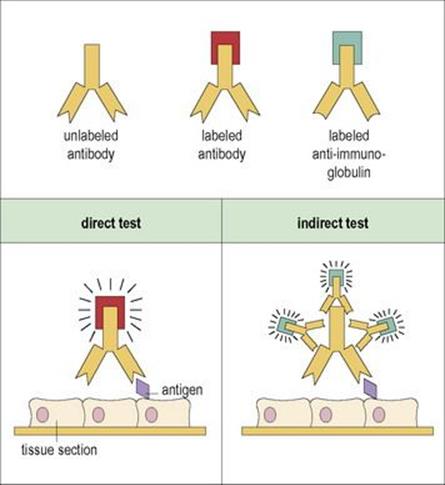
Figure 31.7 The fluorescent antibody test for detection and identification of microbial (or tissue) antigens or antibodies directed against them. In the direct test, antibody labelled with a fluorescent dye is applied to a tissue section bearing the antigen, unbound antibody is washed away, and the bound antibody showing the presence and location of the antigen is visualized by fluorescence microscopy. In the indirect test, antigen is revealed by successive treatments with unlabelled antigen-specific antibody and then fluorescent-labelled anti-immunoglobulin which amplifies the signal (thus if the first antibody is human, the labelled antibody will be an anti-human Ig).
Electron microscopy
The specimen needs to be cut into thin sections for electron microscopy
The electron microscope uses a beam of electrons instead of light, and magnets are used to focus the beam instead of the lenses used in a light microscope. The whole system is operated under a high vacuum. Electron beams penetrate poorly, and a single microbial cell is too thick to be viewed directly. To overcome this, the specimen is fixed and mounted in plastic and cut into thin sections, which are examined individually. Electron-dense stains such as osmium tetroxide, uranyl acetate or glutaraldehyde are applied to the specimen to improve contrast. The electrons pass through the section and produce an image on a fluorescent screen. Images are photographed and enlarged so that the original specimen is magnified many thousandfold (Fig. 31.8).
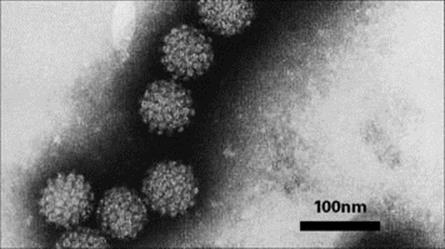
Figure 31.8 Electron micrograph of papillomavirus, the human wart virus.
(Courtesy of the Regional Virus Laboratory, Birmingham, UK.)
Although not routinely used in the clinical laboratory, electron microscopy can aid in the identification of virus particles
Direct examination of specimens allows rapid identification of virus particles and detection of viruses that are difficult or impossible to cultivate (e.g. rotaviruses). Fluid for examination is dried onto a copper grid and examined. About one million virus particles per millilitre are needed if they are to be detectable. The sensitivity can be increased by reacting the fluid with antiviral antibody so that clumps of virus particles are visible. This is known as immunoelectron microscopy, a technique analogous to immunofluorescence in light microscopy.
Detection of microbial antigens in specimens
Detection of specific microbial antigens can be a more rapid method for detecting the presence of an organism than attempting to grow and identify the microbe. The methods include:
• those that detect antigens by their interaction with specific antibodies
• those that detect microbial toxins.
They are summarized in Box 31.3. Detection of microbial genes using DNA probes is discussed later in this chapter.
![]()
Box 31.3 Non-cultural Techniques for Detection of Microbial Products
Non-specific techniques for detection of microbial products
Fatty acid end-products of metabolism of anaerobes can be detected in fluid specimens (e.g. pus, blood) by gas liquid chromatography.
Antigen detection
Detection of soluble carbohydrate antigens by agglutination of antibody-coated latex particles or red blood cells (see Fig. 31.9) e.g.:
• Streptococcus pneumoniae capsule in CSF and urine
• Haemophilus influenzae type b capsule in CSF and urine
• Neisseria meningitidis capsule in CSF and urine
• Cryptococcus neoformans capsule in CSF and urine
• Strep. pyogenes group antigen in throat swabs.
Detection of particular antigens by binding to antibodies labelled with:
• Enzymes (see Fig. 31.11), e.g. ELISA for hepatitis B, rotavirus
• Fluorescent molecules (see Fig. 31.7)
Toxin detection
Detection of exotoxins
• Clostridium botulinum toxin by injection of patient’s serum into mice (unprotected and protected with specific antiserum)
• Clostridium difficile cytotoxin in faeces by addition of suspension to cell culture
• Clostridium perfringens and Staphylococcus aureus enterotoxins in faeces by agglutination of antitoxin-coated latex particles
• Escherichia coli enterotoxin detected by tissue culture or animal model.
Detection of endotoxin
• Endotoxin from cell walls of Gram-negative bacteria detected by Limulus lysate assay conventionally tested by clotting of amoebocyte extracts of the horseshoe (Limulus) crab but also by colorimetric and turbidimetric assays.
Identification of specific microbial products can be a more rapid method for detecting microorganisms than isolation and culture. The available techniques vary in their specificity. Toxins may be detected either by virtue of their antigenic properties or by demonstrating their action. CSF, cerebrospinal fluid; ELISA, enzyme-linked immunosorbent assay. Alternatively, molecular methods such as PCR (see Fig. 31.12) may be used to assess the potential of microorganisms to produce specific microbial products (e.g. toxins) by detecting the presence of their respective genes.
![]()
Specific antibody coated onto latex particles will react with the organism or its product, resulting in visible clumping
For example, the common causative agents of bacterial meningitis (Streptococcus pneumoniae, Haemophilus influenzae and Neisseria meningitidis types A and C) can be detected in CSF by mixing the specimen with specific antibody coated onto latex particles. If the antigen (i.e. the organism or its product) is present, the particles will clump together (Fig. 31.9). These tests give results within minutes of receipt of the specimen, but their sensitivity is not significantly greater than that of the Gram stain, and false-positive results may occur because of cross-reacting antigens. However, they can be a useful diagnostic aid when the patient has received antibiotics and organisms may appear morphologically unidentifiable in the CSF and fail to grow in culture.
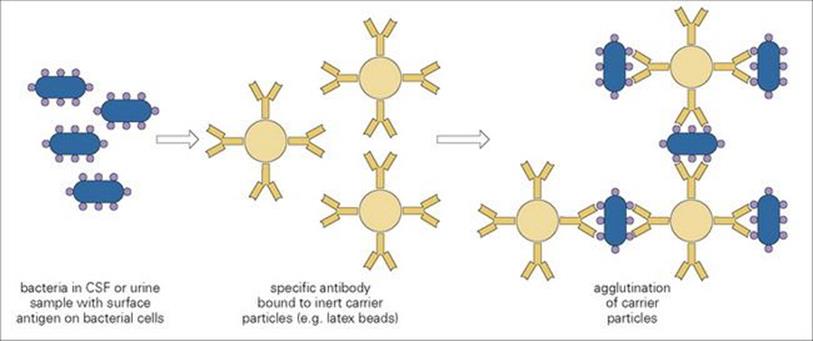
Figure 31.9 When a specimen of cerebrospinal fluid (CSF) containing bacteria (e.g. Haemophilus influenzae) is mixed with a suspension of latex particles coated with specific antibody (e.g. H. influenzae anticapsular antibodies), the interaction between antigen and antibody causes an immediate agglutination of particles, which is visible to the naked eye.
Immunoassay can be used to measure antigen concentration
Antigens can be measured by their binding to a standard amount of antibody, the fractional occupancy of the available antibody binding sites being a measure of the antigen concentration (Fig. 31.10). Usually, the antibody is adsorbed for convenience to a solid phase and the amount of antigen bound assessed using a second antibody labelled with an enzyme which acts on a substance to produce a colour or luminescence (Fig. 31.11) or a fluorescent probe.
• The test employing an enzyme label is referred to as an enzyme-linked immunosorbent assay (ELISA).
• The use of chemiluminescent or time-resolved fluorescent labels gives assays of very high sensitivity.
Earlier forms of immunoassay used labelling with a radioisotope rather than an enzyme or a fluorescent probe.
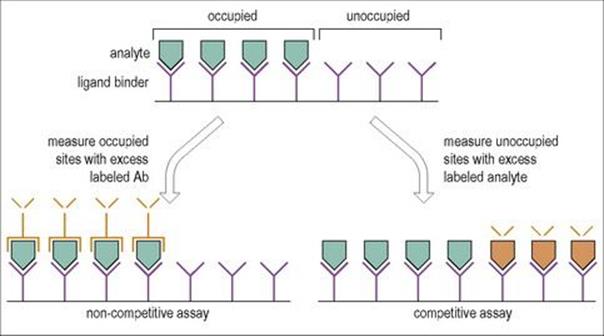
Figure 31.10 The principle of ligand-binding assays. The ligand-binding agent may be in the soluble phase or bound to a solid support as shown, the advantage of the latter being the ease of separation of bound from free analyte. After exposure to analyte, the fractional occupancy of the ligand-binding sites can be determined by competitive or non-competitive assays using labelled reagents (in orange) as shown. In principle, non-competitive assays are more sensitive.
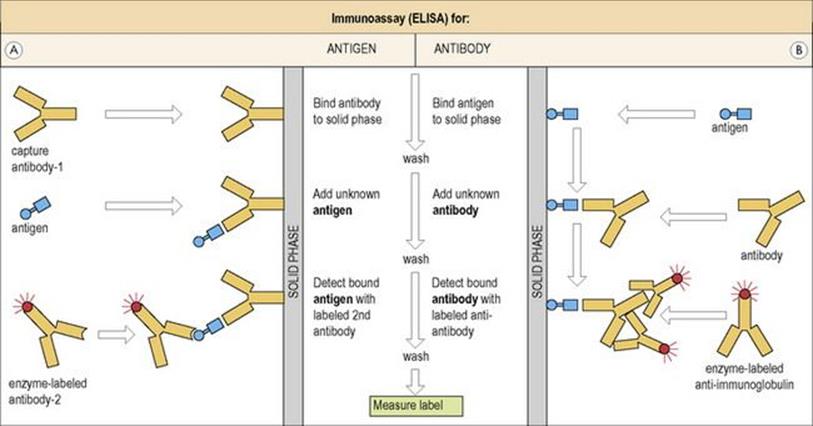
Figure 31.11 Immunoassay on a solid phase (enzyme-linked immunosorbent assay, ELISA). (A) The test antigen is added to solid-phase antibody-1 and the occupancy (see Fig. 31.10) measured by adding an enzyme-labelled second antibody and reading bound enzyme (e.g. peroxidase or alkaline phosphatase) through a colorimetric or luminometric reaction. In some cases, particularly with small antigens, unoccupied sites can be detected by adding a standard amount of labelled antigen. (B) Antibody to be tested is added to solid-phase antigen and is detected by addition of an enzyme-labelled anti-immunoglobulin. (Compare with the indirect test in Figure 31.7, which uses a fluorescent anti-immunoglobulin to detect bound antibody. Similarly, the label in the above assays can be a fluorescent probe rather than an enzyme.)
With modern techniques, multiple assays can be performed on single samples (see below and Fig. 31.19).
Monoclonal antibodies can distinguish between species and between strains of the same species on the basis of antigenic differences
Hybridomas produced by the fusion of ‘immortal’ B-cell tumours and individual normal antibody-producing cells provide a copious source of monoclonal antibodies, all with identical specificities for their relevant antigen. These monoclonal antibodies can be used as diagnostic tools. In direct ELISA (see above), enzyme-conjugated monoclonal antibodies are frequently employed to detect antigens in specimens from patients. Rotaviruses, HIV, hepatitis B virus, herpesvirus and respiratory syncytial virus (RSV) can all be detected directly with monoclonal antibodies in ELISAs. Chlamydia trachomatis infection can be diagnosed within a few hours by a direct fluorescent antibody test employing a monoclonal antibody labelled with fluorescein (see Fig. 31.7; Ch. 21).
Detection of microbes by probing for their genes
Organisms carrying genes for virulence factors can be detected by nucleic acid probes for the virulence factors
A gene probe is a nucleic acid molecule that, when in the single-stranded state and labelled, can be used to detect a complementary sequence of DNA by hybridizing to it. Polynucleotide probes may be obtained from naturally occurring DNA by cloning DNA fragments into appropriate plasmid vectors and then isolating the cloned DNA. However, more typically the sequence of the gene of interest is known and oligonucleotide probes can be synthesized or generated by PCR (see below). The single-strand probes are labelled either with a radioactive isotope or with compounds that give fluorescence or colour reactions in suitable conditions (e.g. biotin streptavidin) and hybridized to the extracted microbial nucleic acid which has first been heat-denatured to make single strands and then immobilized onto a nitrocellulose membrane. Such ‘blotting’ techniques are time consuming and prone to contamination in routine use and have now been largely superseded by PCR methods.
Polymerase chain reaction can be used to amplify a specific DNA sequence to produce millions of copies within a few hours
Polymerase chain reaction (PCR) can theoretically, and often in practice does, detect a single gene-target, i.e. a single organism in the sample being analysed (Fig. 31.12). The method is also rapid; results can be obtained within 1–3 hours, depending on the type of technology used. It is particularly useful for diagnostic work in virology, where many pathogens are difficult to culture. Earlier methods required the PCR products to be analysed on agarose gels and, for diagnostic certainty, some form of nucleic acid probe technique to unequivocally identify the target. This added a considerable amount of time to the analysis and has more recently been replaced by real-time PCR. This approach uses the same basic reagents and techniques as the original method, but with the addition of fluorescently labelled sequence-specific probes. These hybridize to the amplified product (amplicon) as it accumulates and allows the reaction to be monitored in real time (hence the name). The amount of fluorescence accumulated during the reaction is directly proportional to the amount of amplicon produced. By including a set of pre-quantified DNA standards, co-amplified during the reaction, the copy number of nucleic acid in the original sample can be estimated. Because there is no need for post-PCR analysis, the reaction tubes do not need to be opened, which reduces the potential for contamination and provides results in as little as 1 hour. If the pathogen’s nucleic acid is in the form of RNA, it must first be converted into complementary DNA (cDNA), before it can be amplified. This is achieved in an enzymatic step using a reverse transcriptase, prior to the PCR (termed RT-PCR).
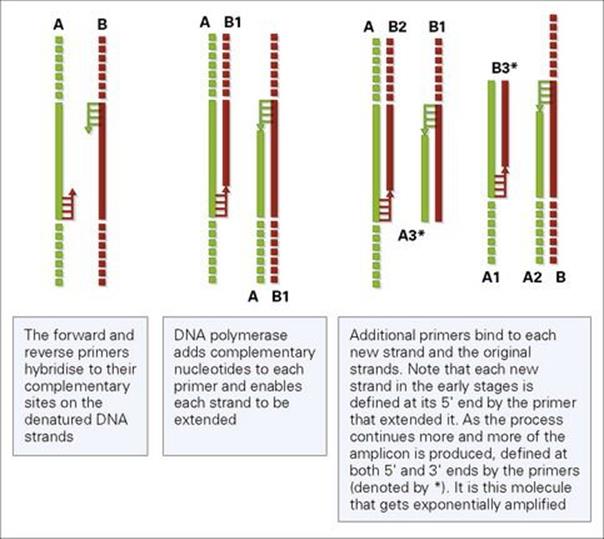
Figure 31.12 The polymerase chain reaction. Short oligonucleotide (ca. 20 bases of DNA) anneal or hybridize to complementary sequences on each DNA strand to be amplified. The strands are separated (denatured) enabling the primers to bind, which are extended by the thermostable polymerase adding complementary nucleotides by repeating the thermal cycling rounds of denaturation, annealing and extension 30–60 times. The original strands to be amplified are shown in the figure as A and B, Subsequent amplified copes are numbered, after early rounds of amplification the desired fragment of DNA to be amplified (amplicon) accumulates and goes on to be copied exponentially.
The specificity of PCR is determined by careful choice of primers
The primers and probes, which are short DNA sequences (ca. 20 nucleotides) must be exact complementary matches to the region requiring amplification and detection. There are now a number of commercial PCR assays available for pathogen detection including C. trachomatis, N. gonorrhoeae, M. tuberculosis and viruses such as cytomegalovirus, hepatitis C virus, herpes simplex virus and HIV (Fig. 31.13). Both qualitative and quantitative detection can be carried out (e.g. HIV-1 RNA load). However, many laboratories continue to design their own primers and probes, particularly for new or re-emerging pathogens. To do this the sequence of the target region must be known. This can be obtained either from DNA sequence databases, or for novel pathogens, where the sequence is unknown, by de novo sequencing in the laboratory. The pathogen nucleic acid can be fragmented by enzymatic methods and cloned into a suitable vector and sequenced using commercially available primers that flank the cloning site. For long regions, new primers can be designed based on the initial data obtained from the first round of sequencing. Thus, DNA sequence analysis is becoming an increasingly important tool in the diagnosis and characterization of infectious pathogens.
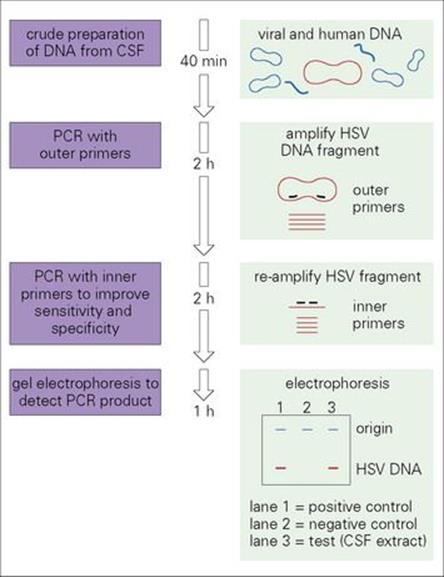
Figure 31.13 Nested polymerase chain reaction (PCR) for the detection of herpes simplex virus (HSV) DNA in cerebrospinal fluid (CSF) from a patient with encephalitis. Nested PCR is a modification of the original PCR technique in which the DNA of interest is amplified first with two primers which recognize sequences some distance apart and then, in a second reaction, with a further pair of primers which recognize sequences within the length of the DNA amplified by the first pair. This technique improves the sensitivity and specificity of PCR.
Dideoxy chain terminator sequencing
This method was developed in the 1970s and, although newer methods are being developed, it is still the cornerstone of routine sequencing technology (Fig. 31.14). The reaction is similar to PCR; a DNA polymerase is used to make copies of the nucleic acid to be sequenced. However, in addition to the four standard nucleotide bases, four dideoxynucleotide base analogues are also used. Dideoxynucleotides lack the 3′-OH group on the sugar moiety necessary for extending the DNA molecule, when one of these molecules is incorporated into the growing chain, extension is terminated, hence the name. Each of the four deoxynucleotides is labelled with a different fluorescent dye, each of which emits light at a different wavelength. The fragments are separated according to their lengths by electrophoresis through a polyacrylamide matrix contained within a capillary tube. A laser excites the different fluorescent dyes and the resulting different wavelengths emitted are picked up by a detector and the sequence of the original nucleic acid is read as a series of fluorescent peaks.
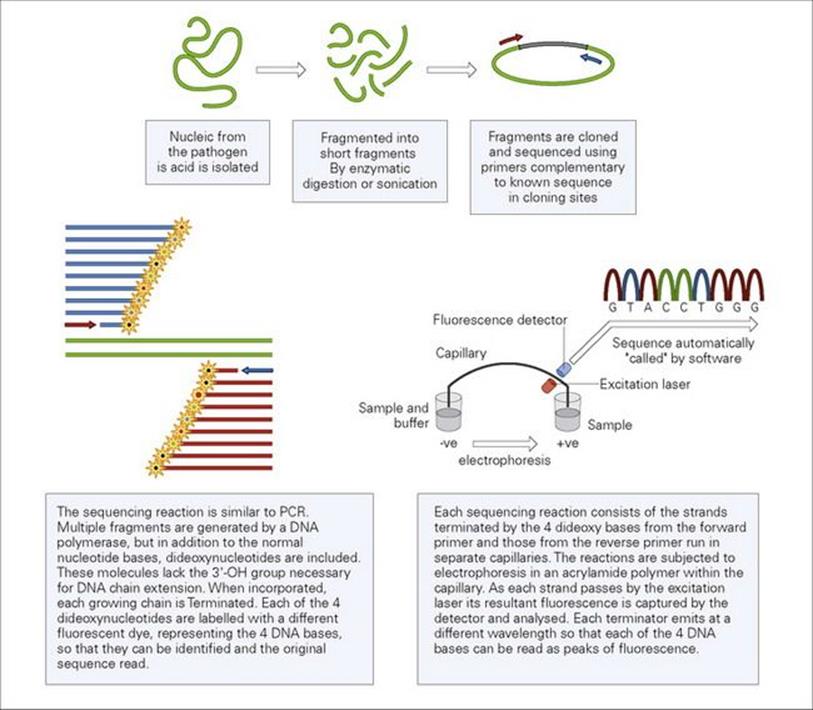
Figure 31.14 Dideoxy capillary sequencing.
Cultivation (culture) of microorganisms
Bacteria and fungi can be cultured on solid nutrient or liquid media
While cultures can be made in liquid media (broth), it is not possible to tell whether there is more than one species present. Therefore, solid media are more useful in diagnostic microbiology. Bacteria and fungi grow on the surface of solid nutrient media (agar-based) to produce colonies composed of thousands of cells derived from a single cell implanted on the surface. Colonies of different species often have characteristic appearances, which can give a clue to their likely identity (Fig. 31.15).
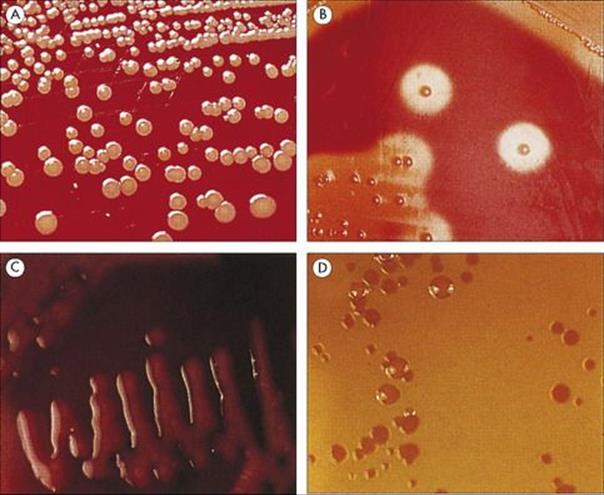
Figure 31.15 Bacterial colonies. A bacterial cell implanted on a solid nutrient medium will multiply to produce a colony containing millions of cells. Different species produce characteristically different colonies, and this feature can be used as a preliminary clue to the identity of the organism. (A) Golden colonies of Staphylococcus aureus. (B) Additional features such as the ability to lyse red blood cells can be demonstrated by culturing bacteria on blood-containing media. Here, β-haemolysis (complete haemolysis) is produced by Streptococcus pyogenes on horse blood agar. (C) Culture media can be made selective by including agents that are inhibitory to some species. For example, MacConkey agar contains bile salts so only those organisms tolerant to bile will grow. In addition, it contains lactose and a pH indicator. Species that ferment lactose change the indicator to bright pink. (D) Non-lactose-fermenting species, such as Salmonella and Shigella, form yellowish colonies.
Different species of bacteria and fungi have different growth requirements
It is possible to grow the majority of species of bacteria and fungi of medical importance in artificial media in the laboratory, but there is no one universal culture medium that will support the growth of them all, and there are still some species that can only be grown in experimental animals (e.g. Mycobacterium leprae and Treponema pallidum). Some bacteria that cannot be cultivated on artificial media (e.g. chlamydia and rickettsia) can be grown in cell cultures (see below).
Many culture media are designed not only to support the growth of the desired organisms, but also to inhibit the growth of others (i.e. they are ‘selective media’).
Specimens collected from body sites that have a normal commensal flora will contain a mixture of organisms from which the pathogen has to be recognized. Specimens are ‘plated out’ on a carefully chosen range of nutrient and selective media to produce single colonies to insure a pure culture. These are subcultured to fresh media for identification and antibiotic susceptibility tests (see below), a procedure which can take 48 h or longer by conventional (non-molecular) approaches (see Fig. 31.1).
Parasites such as Leishmania, Trypanosoma and Trichomonas can be cultivated in liquid media to allow small numbers present in the original specimen (e.g. blood or vaginal secretions) to multiply and thus become easier to detect by microscopic examination. Parasites do not form colonies on solid media in the same way as bacteria and fungi.
Viruses, chlamydia and rickettsia must be grown in cell or tissue cultures
This is because these organisms are incapable of a free-living existence. Most cell cultures used in the diagnostic laboratory are continuous cell lines – human or animal cells adapted to growth in vitro that can be stored at − 80°C until required. The specimen is introduced into the cell culture medium and the presence of viruses detected by observing the cells for a ‘cytopathic effect’ (CPE).
Cell culture techniques are specialized and labour-intensive, and some viruses either cause no CPE or cause a CPE that takes > 1 week to evolve (e.g. cytomegalovirus, although CMV antigens can be detected after 1–2 days in cells). Therefore, alternative methods such as antigen detection (see above), antibody detection (see below) and PCR-based approaches are important for diagnosis.
Identification of microorganisms grown in culture
Bacteria are identified by simple characteristics and biochemical properties
A preliminary identification of many of the bacteria of medical importance has traditionally been made on the basis of the following few simple characteristics of the cells (Fig. 31.16):
• Gram reaction
• cell morphology (e.g. rod or coccus) and arrangement (e.g. pairs or chains)
• ability to grow under aerobic or anaerobic conditions
• growth requirements (simple or fastidious).
Further identification is made on the basis of biochemical properties such as:
• ability to produce enzymes that can be detected by simple tests
• ability to metabolize sugars oxidatively or fermentatively (aerobically or anaerobically)
• ability to use a range of substrates for growth (e.g. glucose, lactose, sucrose).
While these tests can be done individually (e.g. in broth media containing the specifically required reagents), they are more commonly performed using commercial kits or automated systems which have the potential to give a rapid (e.g. 2–4 h) indication of pathogen identity based on biochemical profiles.
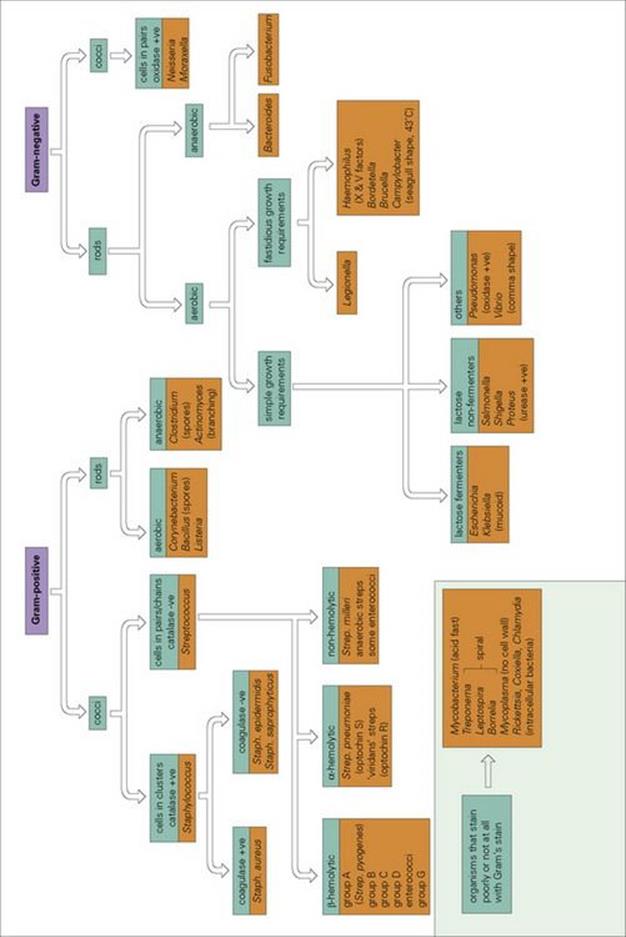
Figure 31.16 Identifying bacteria. The preliminary investigation of the bacteria of medical importance has traditionally been made on the basis of a few key characteristics (see text). Further identification may then be made on the basis of biochemical and serologic tests.
Some species are identified on the basis of their antigens by reacting cell suspensions with specific antisera.
Antibiotic susceptibility can only be determined after the bacteria have been isolated in a pure culture
A variety of methods are available for antimicrobial susceptibility testing, including broth microdilution and automated instrument approaches. However, the most widely employed method assesses antibiotic susceptibility by applying filter paper disks, which contain different antibiotics, onto a lawn of the test organism which has been seeded onto an agar plate (i.e. disk diffusion). During overnight incubation, the organisms grow and multiply and the antibiotics diffuse out from the disks and inhibit growth around the disk. Therefore, after isolation of bacteria from a specimen, a further incubation period (overnight for disk diffusion testing) is required before antibiotic susceptibility results are available. Methods for antibiotic susceptibility tests are described in more detail in Chapter 33.
Fungi are identified by their colonial characteristics and cell morphology
Fungi are identified from colonies or pure cultures largely on the basis of colonial characteristics (e.g. colour) and the morphology of the individual cells viewed under the microscope (Fig. 31.17). Biochemical tests (substrate assimilation) can be used for detailed identification of yeasts of medical importance. In general, fungi grow more slowly than bacteria, and final identification may take up to 2 weeks.
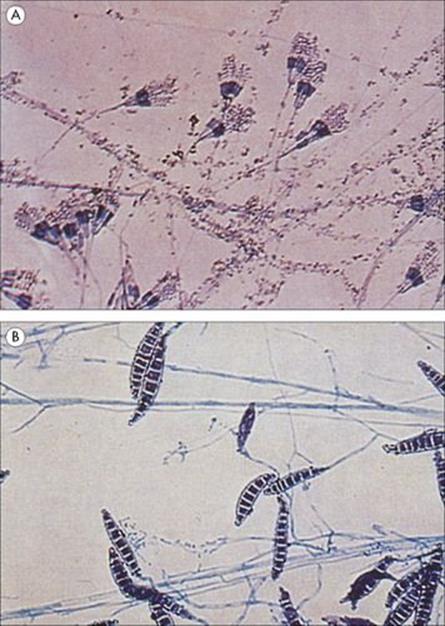
Figure 31.17 Fungi under the microscope. Fungi can be grown on agar culture media in the same way as bacteria, but most species grow much more slowly than bacteria and it may take up to 2 weeks for a colony to form. Colonial characteristics (such as colour) are helpful in the identification of fungi, but confirmation depends upon microscopic examination of the hyphae and sporing structures. (A) Penicillium in a wet preparation showing the conidiophores and free conidia. (B) Macroconidia of Microsporum canis stained with lactophenol cotton blue.
Protozoa and helminths are identified by direct examination although newer molecular methods are also available
Many protozoa and parasites can be identified by direct examination of specimens without resorting to culture, and therefore the results can be obtained on the day of receipt of the specimen in the laboratory:
• Protozoa are traditionally identified on the basis of their morphologic characteristics – different stages of the lifecycle may be visible in different specimens from the same patient and at different stages in the disease (Fig. 31.18)
• Helminths are commonly identified by the macroscopic appearance of the worm (e.g. Ascaris or Enterobius) or by microscopic examination of specimens (e.g. faeces or urine) for eggs of, for example, schistosomes (see Ch. 22).
However, molecular approaches to diagnosis are becoming increasingly more common including PCR detection and differentiation of Entamoeba species, Giardia, and Cryptosporidium.
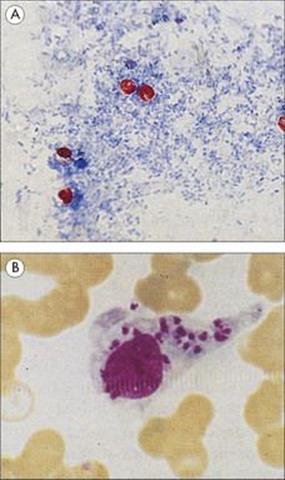
Figure 31.18 Although some parasites can be cultivated in the laboratory, identification is usually based on microscopic appearances in the specimen. (A) Acid-fast stain of Cryptosporidium in faeces. Like mycobacteria, this organism is able to retain the pink carbol fuchsin stain when challenged with acid alcohol. (B) Leishmania donovani (Donovan bodies) in a stained preparation from a specimen of bone marrow.
Viruses are usually identified using serologic tests
Viruses may be identifiable by their cytopathic effect in cell culture and their morphology in electron microscopic preparations (Fig. 31.8). A number of viruses may now be identified by nucleic acid-based tests (e.g. probes and PCR; see above), but diagnosis is also often made by detecting viral antigens or by testing for the presence of specific antibodies in the patient’s serum (see below).
Mass spectrometry heralds a novel diagnostic era
One of the most promising new approaches to the identification of bacteria and fungi involves the use of mass spectrometry or, more specifically, matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI-TOF). As MALDI-TOF equipment becomes more widely available, it is being employed increasingly for the identification of microbial pathogens through analysis of their predominant mass spectral protein fingerprints. The field is being heavily researched and the potential value of identifying a variety of smaller molecules is also being explored with the promise of exciting new developments in the future.
Antibody detection methods for the diagnosis of infection
Serologic tests (the study of antigen–antibody interactions) are used:
• to diagnose infections
• to identify microorganisms (see above)
• to type blood for blood banks and tissues for transplantation.
Diagnoses based on detecting antibodies in patients’ sera are retrospective
The major disadvantage of a diagnosis based on the detection of antibodies in a patient’s serum is that it is retrospective, as 2–4 weeks must elapse before IgG antibodies produced in response to the infection are detectable. What is more, a positive result indicates only that the patient has come into contact with the infection at some time in the past. However, IgM antibodies are detected earlier in the infection (7–10 days) and are usually indicative of active, as opposed to past, infection. It may also help to show that the patient has ‘seroconverted’ by demonstrating a fourfold or greater rise in antibody titre between sera collected in the acute and convalescent phases of the disease.
Antibody detection can be invaluable for identifying organisms that grow either slowly or with difficulty
Despite the drawbacks mentioned above, antibody detection is the main method for the laboratory diagnosis of viral infections. The techniques employed often allow several different infections to be screened for simultaneously (e.g. causes of atypical pneumonia, see Ch. 19). Sera should be collected during the acute phase of the disease and stored at − 20°C until a convalescent-phase serum is available; the two sera are then tested in parallel. Few diagnoses can be made with any confidence on the results of single serum samples, but sometimes early testing is justified if there is a clinical suspicion of a rare infection that the patient is unlikely to have encountered before (e.g. legionellosis). Previous immunization makes it difficult if not impossible to interpret some serologic tests, because antibodies detected may be the result of immunization or infection (e.g. the Widal test for the serologic diagnosis of enteric fever, see Ch. 22).
Common serologic tests used in the laboratory to diagnose infection
Solid-phase immunoassays can be used to estimate antibody in a given sample
These assays have been described previously (see Fig. 31.11). The amount of antibody binding to the solid-phase antigen is a measure of the antibody content of the original sample, and can be detected by adding a second antibody conjugated with a fluorochrome or an enzyme (e.g. phosphatase or peroxidase) that produces a colour or luminescent reaction with a given substrate.
Modern techniques permit the simultaneous assay of several analytes in the same sample. Examples of two current multiplexing technologies are presented in Figure 31.19.

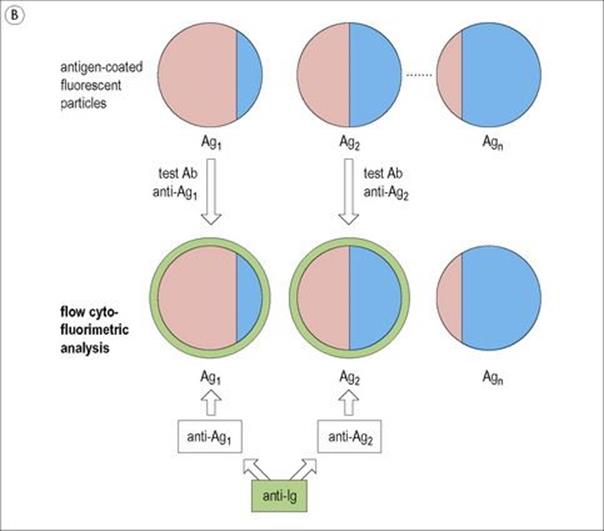
Figure 31.19 Multiplex (i.e. multiple) solid-phase immunoassays on single test samples. (A) A diagnostic antigen microarray permitting the assay for multiple antibodies within each individual serum sample tested on a 5 × 5 array of antigen dots in each well of a membrane-based microtitre plate using a luminescent enzyme-linked anti-immunoglobulin readout. Twenty of the dots in each well simultaneously evaluate 10 markers in duplicate with 5 dots being used as controls, two of which give strong luminescence signals in two central control spots in all wells, the other 3 in the equatorial row being buffer spots used for the background subtraction value. Varied intensities of other spots correlate with the level of antibodies present in each patient’s serum (Courtesy of J. McBride). (B) Luminex system in which a variety of particles, each bearing single antigens on their surface, are identified by their individual ratio of two different fluorochromes using flow cytometry (see Fig. 31.25). When antibodies in the test serum bind to a particle, they can be revealed by coating with an anti-immunoglobulin bearing a third fluorochrome. In this way, antibodies to several different antigens within a given test sample, of blood for instance, can be quantitatively assayed.
A variety of tests assess the ability of antibodies to inhibit microbial activity
A number of tests focus on the ability of antibodies in a patient’s serum to inhibit some biologic faculty of the microorganism in question. An example is the anti-streptolysin O test, in which the streptolysin O toxin is neutralized by antibody. The extent to which the test serum can be diluted before it fails to prevent the toxin from lysing red cells provides a convenient titre (Fig. 31.20). The ability of a patient’s serum containing specific antibody to immobilize motile bacteria – for example, the Treponema pallidum inhibition (TPI) test – is another example.
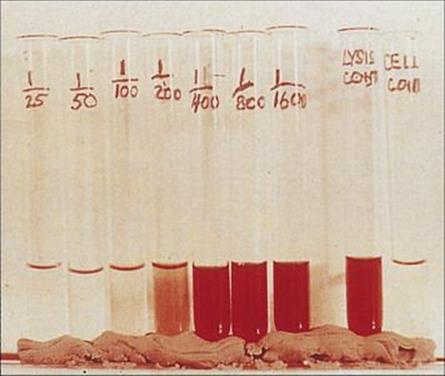
Figure 31.20 Illustration of the anti-streptolysin O (ASO) test which is now performed primarily by automated instrumentation. The O-toxin lyses red cells. Test serum is diluted until the antibodies it contains it no longer inhibit lysis by a standard concentration of toxin. Positive and negative controls are included in the test (right).
Antibodies can also mask viral molecules such as the influenza haemagglutinins, which are involved in specific adherence to cells, allowing the development of a haemagglutination inhibition test (Fig. 31.21). This assay is a classic technique only in use in reference laboratories.
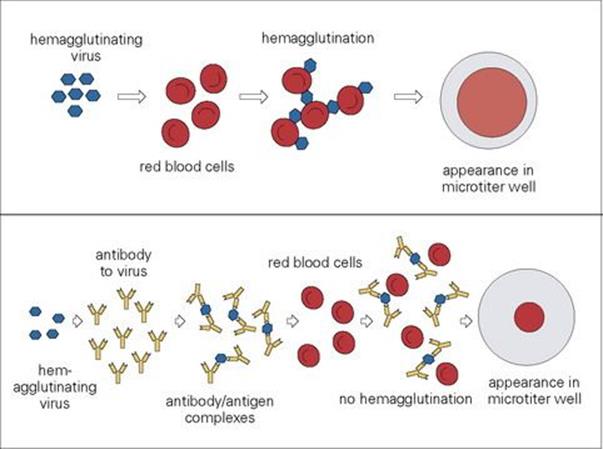
Figure 31.21 Haemagglutination inhibition. Some viruses (e.g. influenza) have haemagglutinin molecules on their outer surface, and when virus particles are mixed with red blood cells, they cause haemagglutination. In the presence of specific antibody, however, haemagglutination is inhibited. This test can therefore be used to detect the presence of antibodies to influenza virus in a patient’s serum. Note that the routine test for influenza is PCR but that haemagglutination inhibition is used as a confirmatory test by the reference laboratory.
Antibodies to cytopathic viruses can be detected by the ability of the patient’s serum to prevent virus infectivity
In the case of cytopathic viruses, antibodies can be detected by the ability of the patient’s serum to prevent the development of a cytopathic effect. These antibodies are called neutralizing antibodies. The important applications of these methods are referred to in the appropriate systems chapters (see Chs 18–30).
Point of care tests
New simpler diagnostic tests are being developed that can be used at the point of care
For example, clinicians increasingly exploit simple dipsticks to directly measure sugar in urine. Much current research aims to design new diagnostic assays that could be performed by non-laboratory staff in a clinic, surgery, or other point-of-care setting. Using lateral flow technology (similar to that used in pregnancy tests), microorganisms might be detected via antibodies to particular antigens. Such tests are already in use for HIV, although they should be confirmed by a second assay and all doubtful or uncertain results referred to the laboratory. Even more miniaturized ‘lab on a chip’ technology is in development, where reactions take place within a piece of plastic rather than the laboratory.
Assessment of host defence systems
The opsonic activity and activities of individual components of complement can also be assessed
Although assessment of overall serum complement activity is a relatively uncommon procedure at the present time, it is often of value to assess the opsonic activity of complement in the serum sample by measuring its ability to facilitate the uptake of a microbial particle by a phagocytic cell (Fig. 31.22).
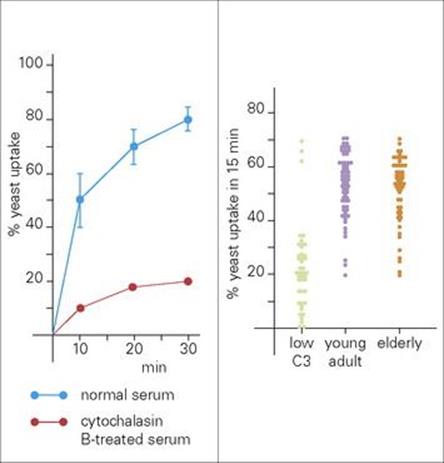
Figure 31.22 Opsonic activity of serum. (A) Time course for the uptake of yeast opsonized with a normal serum by polymorphonuclear neutrophil leukocytes (PMNs) from 12 healthy donors, and uptake of yeast by PMNs from one donor after treatment with cytochalasin B (40 mg/mL), which inhibits phagocytosis. (B) The distribution of opsonic activity for 150 sera from young healthy, elderly and pathologic sera.
(Data from Kerr et al., Clin Exp Immunol 54:793–800, 1983.)
The activities of individual components of the complement system can be evaluated either by:
• their ability to be titrated into a complement-dependent lytic system in which the component to be tested is lacking
• direct immunochemical measurement, often using gel precipitation reactions.
The nitroblue tetrazolium (NBT) test is used to assess phagocytic activity
The ability of neutrophils to become phagocytic and to concurrently reduce molecular oxygen can be assayed by the nitroblue tetrazolium (NBT) test. When yellow NBT dye is added to blood, it forms complexes with heparin or fibrinogen in the sample. These complexes are then phagocytosed by neutrophils that have been activated by the addition of exogenous endotoxin. The dye complex is taken into the stimulated neutrophils and substitutes for oxygen by acting as a substrate for the reduction process, forming as a result a blue insoluble formazan (Fig. 31.23).
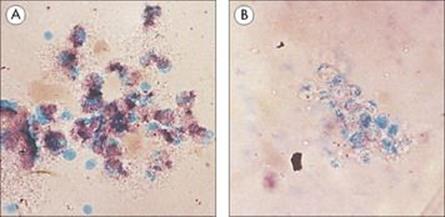
Figure 31.23 Nitroblue tetrazolium (NBT) test. In normal polymorphs and monocytes, reactive oxygen intermediates (ROIs) are activated by phagocytosis, and yellow NBT is converted to purple-blue formazan (A). Patients with chronic granulomatous disease (CGD) cannot form ROIs and so the dye stays yellow (B).
(Courtesy of A.R. Hayward.)
Lymphocytes
The development of T-effector cells to an antigen can often be revealed by intradermal challenge with that antigen. Such an intradermal challenge usually gives rise to erythema and induration, peaking at around 48 h (Fig. 31.24). This time course has led to the reaction being described as ‘delayed-type hypersensitivity’, and is the basis of the Mantoux skin test for tuberculosis (see Chapter 19).
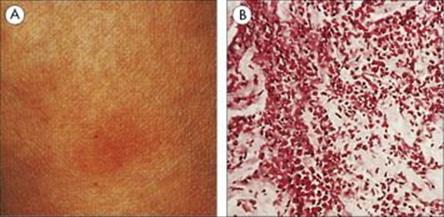
Figure 31.24 Tuberculin-type delayed sensitivity. The dermal response to antigens of leprosy bacillus in a sensitive subject (the Fernandez reaction) is characterized by (A) red induration maximal at 48–72 h and (B) dense infiltration of the injection site with lymphocytes and macrophages. (H&E, × 80.)
Overall responsiveness of the T-cell population can be probed by using materials such as phytohaemagglutinin or concanavalin A, which are polyclonal stimulators in the sense that they activate T-cell populations independently of their precise antigen specificity. However, when peripheral blood cells are incubated with antigen in vitro, the specifically sensitized T cells, which represent only a very small fraction of the total, become activated and divide. Examination of the cultures will reveal blast cells and mitotic divisions, but the response can also be detected by the incorporation of radiolabelled thymidine, or of a fluorescent dye, such as CFSE, which provides a measure of cell proliferation.
Lymphocytes are counted and classified by detecting their cell surface molecules
Lymphocyte differentiation is accompanied by the expression of related molecules on the cell surface. Detection of these molecules by immunofluorescent techniques allows their enumeration and, in addition, their classification into different subpopulations (Fig. 31.25). Monoclonal antibodies are widely used to define these differentiation molecules, using flow cytofluorimetry (Fig. 31.26), which is a more rapid and less laborious means of analysing lymphocyte subpopulations than conventional fluorescent microscopy. The fluorescence-activated cell sorter (FACS) separates subpopulations delineated by their cytofluorimetric parameters. Newer FACS machines can measure 10–18 different fluorescent labels simultaneously, allowing both the surface phenotype of the cells and its function to be assessed.
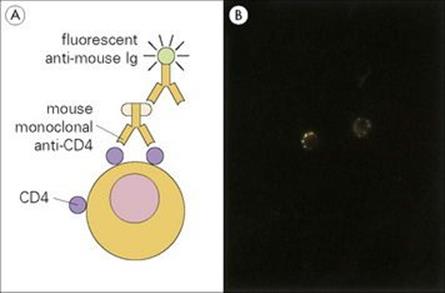
Figure 31.25 Visualization of lymphocyte surface differentiation molecules by immunofluorescence. (A) The double antibody test using mouse monoclonal antibodies to the required surface molecule. Another example of the indirect fluorescent antibody test (Fig. 31.7). (B) Direct demonstration of immunoglobulin receptors on the surface of two B lymphocytes by fluorescent anti-immunoglobulin. Aggregation and capping of the immunoglobulin surface receptors by the anti-immunoglobulin reagent is evident.
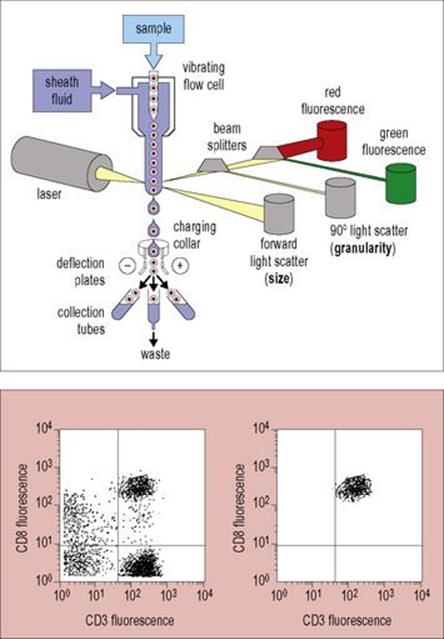
Figure 31.26 Flow cytofluorimetry. Cells in the sample are stained with specific fluorescent reagents to detect surface molecules and then stream one at a time past a laser. Each cell is measured for size (forward light scatter) and granularity (90° light scatter), as well as for red and green fluorescence, to detect two different peripheral blood surface markers, in this instance, CD8 and CD3, respectively (but modern instruments can detect many more different fluorophores). In a cell sorter, the flow chamber vibrates the cell stream, causing it to break into droplets which are then charged according to an arbitrary cut-off ‘gate’ and can then be steered by deflection plates under computer control to collect different cell populations according to the parameters measured. In the example shown in the left panel, four populations can be seen and after appropriate gating, the CD8 population in the right upper quadrant can be selected; reanalysis gives the plot seen on the lower left panel.
(Redrawn from Male D, Brostoff J, Roth DB, Roitt I. Immunology, 7th edition, 2006. Mosby Elsevier, with permission.)
Individual cells secreting antibodies or cytokines can be counted by the ELISPOT technique or by flow cytometry
The lymphocytes are incubated on a membrane impregnated with antigen, for antibody detection or anticytokine monoclonal antibody to detect cytokines (Fig. 31.27). The secreted product is identified by conventional ELISA-type readout.
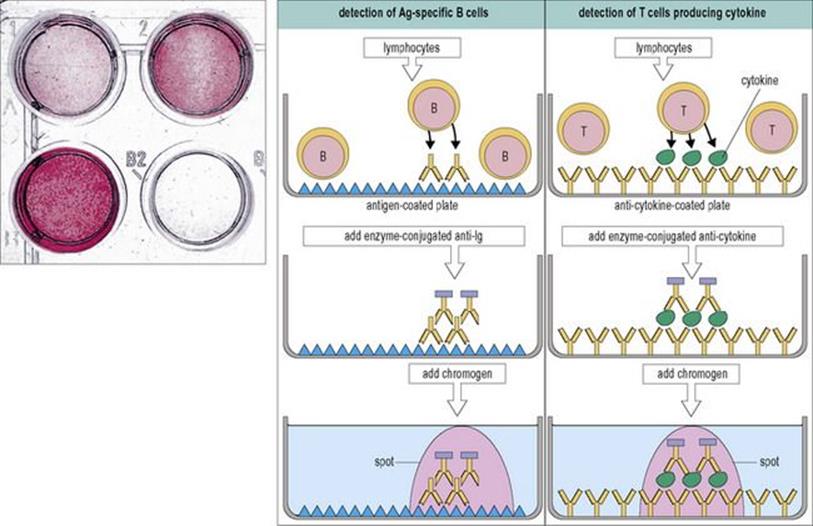
Figure 31.27 The ELISPOT assay for counting lymphocytes secreting antibodies or cytokines. The secreted products (antibodies from B cells and cytokines from T cells) are bound by the solid-phase capture molecules immediately beneath the cell and revealed by a colour reagent as a spot corresponding with the secreting cell. Wells with different numbers of ELISPOTs are shown top left.
(Redrawn from Male D, Brostoff J, Roth DB, Roitt I. Immunology, 7th edition, 2006. Mosby Elsevier, with permission.)
Recently, two alternative approaches have been developed. One utilizes inhibitors of cytokine export (e.g. metabolic poisons such as brefeldin A that trap cytokines within the endoplasmic reticulum) to block cytokine secretion so that these molecules can be immunostained after cellular permeabilization. Cells can then be stained for intracellular cytokines using specific antibodies, followed by flow cytometric analysis, as described earlier. The other approach makes use of bispecific antibodies that can simultaneously bind to a T-cell surface marker (such as CD4) while the other Fab arm is specific for a cytokine. The cytokines are captured as they are secreted from cells but, due to the bispecific nature of the antibody, the cytokine becomes stably attached to the cell making it and can then be detected with a different cytokine-specific antibody conjugated to a fluorochrome, again using a flow cytometer.
The ability of cytotoxic T cells to attack targets is conventionally assayed using a radioisotope
Cytotoxic T cells attack targets such as virally infected cells, and this ability is conventionally assayed by prelabelling the target with a radioisotope such as 51Cr, and then looking for release of the isotope into the supernatant from damaged cells (Fig. 31.28). Cytotoxicity can also be measured by non-radioactive techniques using dyes.
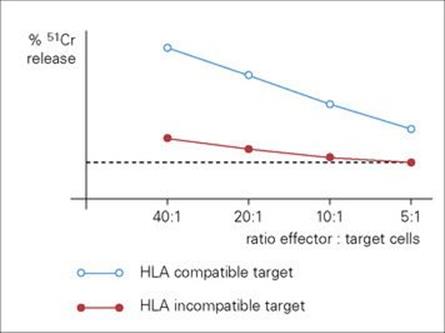
Figure 31.28 Measurement of cytotoxic activity of human lymphocytes against influenza-infected target cells. Only those targets that share human leukocyte antigens (HLA) haplotype with the cytotoxic cell donor are attacked (haplotype restriction) with consequent release of 51Cr. The dotted line indicates background release of isotype from target cells incubated in the absence of effector cells.
Putting it all together: detection, diagnosis, and epidemiology
As seen more fully in Chapter 32, understanding the epidemiology of an infection can help to define the correct strategies for control at the population level. However, this understanding, and decisions about control, both depend heavily upon the ability to recognize outbreaks of disease, to follow their progress and to identify the causative organism concerned. Detection and diagnosis are therefore key activities here, as they are for treatment of infection at the level of the individual.
Descriptive epidemiology involves asking questions about an outbreak of disease that will help to identify the pathogen and the source of infection. It is important to have a case definition, which includes the symptoms of the disease as well as details of the individuals involved and the timing of events. Analysis of these data should make it possible to say where and how the outbreak has arisen, who is at risk and what treatment is necessary to control further infection (see Box 31.4 on Legionnaires’ disease for an example). Measures used may involve antibiotic treatment of those immediately affected, or vaccination if a large number are at risk (e.g. meningitis outbreaks in university students). For sexually transmitted disease (see above) an important element of detection is to establish contact patterns, or mixing matrices, so that individuals who may acquire an infection can be treated and further transmission prevented.
![]()
Box 31.4 Lessons in Microbiology
Legionnaires’ disease – a case study
Background
War veterans of the Pennsylvania American Legion held their convention at a hotel in Philadelphia on 21–24 July 1976. By early August, an outbreak of severe pneumonia (cause unknown) among participants was reported. Deaths occurred despite antibiotic treatment.
Case definition
• Attendance at the convention or presence at the hotel between 1 July and 18 August
• Onset between those dates of cough, fever, verified pneumonia.
A total of 182 patients met this definition, of whom 149 had attended the convention and nine had been at the hotel for other conventions in the time period. An additional 39 patients had the clinical condition, but had not been in the hotel. They had, however, been within one block of the hotel in the relevant time period.
The epidemic
Cases appeared in late July, peaking between the 25th and 27th. A total of 78% of the cases were male, most were older than 50. The incubation period was between 2 and 10 days. A significant proportion of cases had spent time in the lobby or stood outside the hotel to watch the parade. There was no significant person-to-person spread.
Conclusion
The evidence pointed to an air-borne infectious agent, most likely acquired in the hotel lobby or immediately outside, entering the body via respiration. Initially, although the clinical evidence suggested a bacterial infection (large numbers of neutrophils in the sputum), no organisms could be demonstrated. Legionella, a previously unknown bacterium, was isolated and identified shortly afterwards. Erythromycin was found to be effective. The biology of Legionella pointed to control through disinfection and high-temperature treatment of water supplies and air-conditioning plant.
![]()
This approach to outbreaks of known or new disease follows their chance discovery as a result of clinical observations, exemplified by the discovery of AIDS in 1981 through the increased occurrence of Pneumocystis carinii (now known as Pneumocystis jirovecii) infection and of Kaposi’s sarcoma in homosexual males. A more systematic approach to detection relies on a regular notification system – a surveillance system that routinely records episodes of a number of legally notifiable diseases. Such systems operate nationally through government or federal health organizations, as well as internationally through the World Health Organization for diseases such as cholera, yellow fever and plague. Regular monitoring of this kind makes it easier to identify outbreaks, because it provides the baseline against which ‘the occurrence of cases in excess of expectancy’ (the definition of an epidemic) can be measured.
Once outbreaks of infectious disease have been detected, the pathogen concerned can be identified by conventional diagnostic procedures, to ensure that the appropriate antibiotic or vaccination is given.
![]()
 Key Facts
Key Facts
• Microbiologic confirmation of a clinical diagnosis of infection depends upon the collection of high-quality specimens and their rapid despatch to the laboratory with all the necessary supporting information.
• Laboratory tests detect microorganisms or their products or evidence of a patient’s immune response to infection.
• While coming from different perspectives, culture and serologic methods are important, cooperative approaches to the identification of clinically important pathogens.
• Newer molecular techniques (e.g. involving PCR and mass spectroscopy) are increasingly used to detect pathogens rapidly; however, antimicrobial susceptibility can only be determined and appropriate treatment information provided by isolating organisms in pure culture.
• Growth of bacteria requires at least 18 h (isolation of viruses and of fungi may take much longer); therefore standard culture results cannot be expected in less than 24 h although newer diagnostic tests are more rapid.
• Interpretation of culture results depends upon the source of the specimen. From sites that are normally sterile, any isolated organism is significant. From sites colonized by commensal flora, isolating and identifying the pathogen can be more difficult.
• Good communication between the clinician and the microbiologist is extremely important.
![]()