Organic Chemistry I For Dummies, 2nd Edition (2014)
Part III. Functional Groups
Chapter 14. Side-by-Side: Conjugated Alkenes and the Diels–Alder Reaction
IN THIS CHAPTER
Seeing the difference between conjugated and isolated alkenes
Examining 1,2- and 1,4-additions to alkenes
Distinguishing kinetics from thermodynamics
Seeing the Diels–Alder reaction of conjugated dienes
Some of the first reactions that you saw in organic chemistry were likely the reactions of alkenes (discussed in Chapter 10), but those reactions don’t close the book on this important functional group. Multiple double bonds that alternate in a carbon chain, called conjugated alkenes, have different properties and reactivities than double bonds that exist all by their lonesome. In this chapter, I explore some of these differences, and use the reaction of conjugated double bonds with acids to explain the difference between kinetics and thermodynamics. I also show you perhaps the most interesting reaction in organic chemistry, the Diels–Alder reaction, which lets you easily form rings and bizarre bicyclic structures.
Seeing Conjugated Double Bonds
Conjugated double bonds are molecules in which two or more double bonds are separated by one carbon-carbon single bond, as shown in Figure 14-1. The double bonds are alternating, in other words. Isolated double bonds are those that are separated by more than one carbon-carbon single bond.

FIGURE 14-1: Conjugated versus isolated dienes.
One feature of conjugated alkenes is that they’re more stable than isolated alkenes. You can justify this added stability by writing out the resonance structures for a conjugated alkene (see Chapter 3 for more on resonance structures) and recalling that resonance is a stabilizing feature of molecules. Figure 14-2 shows the resonance structures for the conjugated diene butadiene. In contrast, isolated double bonds have no resonance structures and so are less stable.
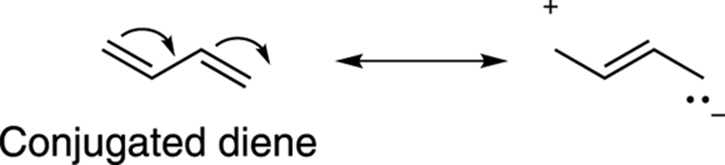
FIGURE 14-2: One resonance structure for butadiene.
Addition of Hydrohalic Acids to Conjugated Alkenes
In addition to their greater stability over isolated double bonds, conjugated double bonds react differently than isolated double bonds. The addition of hydrohalic acids (like HBr, HCl, and so forth) to double bonds is shown in detail in Chapter 10. But hydrohalic acids react somewhat differently with conjugated double bonds than they do with isolated double bonds.
You can see how this difference in reactivity arises by looking at the mechanism for this reaction, shown in Figure 14-3. The first step in the mechanism is the protonation of one of the double bonds to form the carbocation. (Note that in general the cation prefers to be secondary rather than primary; see Chapter 10 for more on carbocations.) This cation has two resonance structures, one in which the positive charge resides on the number-two carbon and one in which the positive charge resides on the number-four carbon. Thus, the true structure of the cation lies somewhere in between these two resonance structures, with a structure that has some positive charge located on the number-two carbon and some positive charge on the number-four carbon. The halide can attack either of these two positively charged spots. Attacking the number-two carbon leads to the 1,2-addition product (because the hydrogen has added to position one and the halide to position two), while attacking at the number-four carbon leads to the 1,4-addition product (because the hydrogen has added to position one and the halide to position four).
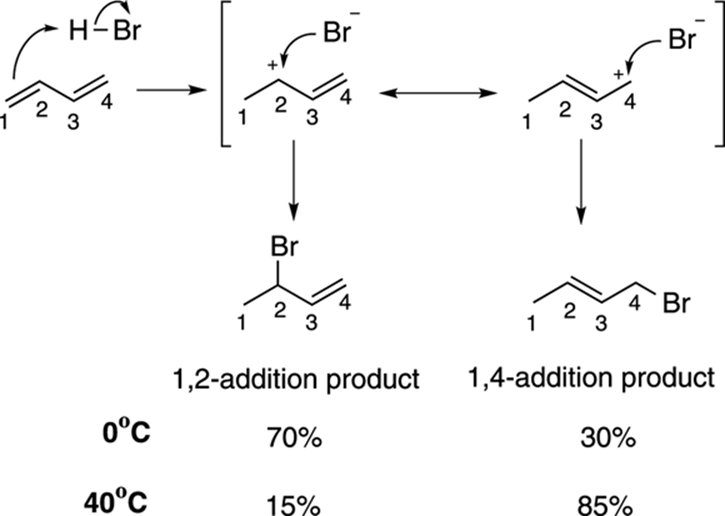
FIGURE 14-3: Mechanism of 1,2-addition and 1,4-addition.
Seeing the reaction diagram of conjugate addition
So, which of the two conjugate addition products will be favored? Interestingly, the relative amount of each material produced depends on the temperature of the reaction. While you get mostly 1,2-addition at lower temperatures (at 0°C), you get mostly 1,4-addition at higher temperatures (at 40°C). To see why this is the case, you need to look at the reaction diagram for the two different pathways, shown in Figure 14-4. Note that a reaction diagram plots the progress of the reaction versus the energy of the reaction (refer to Chapter 12 for more on reaction diagrams).
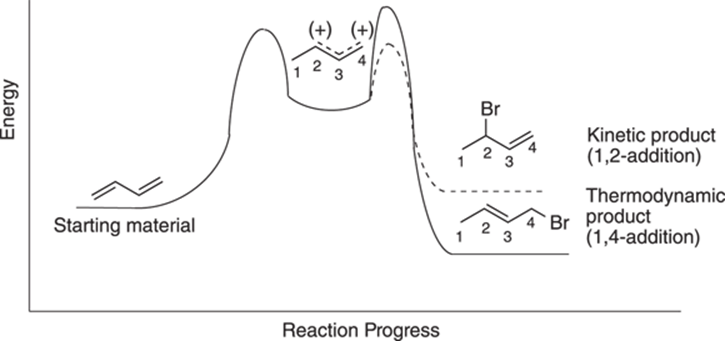
FIGURE 14-4: Reaction diagram for conjugate addition.
Both pathways are initially identical because both go through the same carbocation. Then the pathways diverge. You can see from the reaction diagram that the 1,2-addition product has a lower energy of activation for the second step (a smaller energy hill to climb), but gives a higher-energy product, while the 1,4-addition reaction pathway has a higher energy of activation for the second step, but leads to a more stable, lower-energy product.
At low temperatures, the reaction supplies enough energy for the reaction to go over the smaller barrier to reach the 1,2-addition product, but not enough energy to go over the higher-energy barrier to reach the 1,4-addition product. Because at low temperatures the reaction is irreversible — that is, after the reaction has gone over the energy hill to make product, the reaction doesn’t have enough energy to climb the higher hill back to the cation intermediate — after the starting material is converted to product, it stays as product and doesn’t go back to the intermediate carbocation. In this way, at low temperatures you favor the 1,2-addition product that has the lower activation barrier, even though it results in a higher-energy product. The product formed with the lower activation barrier is called the kinetic product using organic-speak.
At higher temperatures (around 40°C), the reaction is reversible (has enough energy to return to the cation after it’s made the kinetic product) and can reach equilibrium. Any system at equilibrium will favor the lower-energy product, called the thermodynamic product. The reaction at high temperature has enough energy to go over the higher-energy activation barrier to reach the more stable product. Additionally, because enough heat is present to make the 1,2-addition reaction reversible, eventually most of the 1,2-product will go back up the hill to the carbocation state. Once there, the reaction will eventually lead to 1,4-addition, because this reaction gives the lower energy product.
Comparing kinetics and thermodynamics of conjugate addition
Kinetics and thermodynamics are easy to confuse, but here’s how you can remember the difference between the two. Kinetics refers to the rates of reactions. Reaction rates depend on the heights of the energy barriers in a reaction (the activation energies). Thermodynamics, on the other hand, refers to the relative energies of the starting material and the product (the change in energy). If you want a reaction to be under kinetic control, you run the reaction at low temperature so that only low-energy hills will be climbed by the reaction, and the kinetic product will be favored. At higher temperatures, when a reaction is at equilibrium, the reaction is under thermodynamic control, and the lowest-energy product is favored, regardless of the height of the activation barrier.
 In general, 1,2-addition typically leads to the kinetic product, while 1,4-addition leads to the thermodynamic product (refer to Figure 14-4 for the specific case of the HBr addition to butadiene). So to get the 1,2-addition product on a conjugated alkene you typically run the reaction at lower temperature, and to get the 1,4-addition product you run the reaction at higher temperatures.
In general, 1,2-addition typically leads to the kinetic product, while 1,4-addition leads to the thermodynamic product (refer to Figure 14-4 for the specific case of the HBr addition to butadiene). So to get the 1,2-addition product on a conjugated alkene you typically run the reaction at lower temperature, and to get the 1,4-addition product you run the reaction at higher temperatures.
The Diels–Alder Reaction
One of the most interesting differences between isolated alkenes and conjugated alkenes is that conjugated alkenes can undergo the Diels–Alder reaction. The Diels–Alder reaction is probably the funkiest reaction you encounter in organic chemistry. It’s also one of the most valuable reactions in organic chemistry, because in a single step this reaction produces two valuable carbon-carbon bonds (so valuable to chemists that the discoverers, Otto Diels and Kurt Alder, shared the Nobel Prize in chemistry for discovering this reaction). The reaction is extremely useful in the construction of six-membered rings and bicyclic molecules.
The general form of the Diels–Alder reaction is shown in Figure 14-5. In this reaction, a conjugated diene reacts with an alkene (called the dienophile, or diene lover) to form a six-membered ring. The reaction occurs in a single step; it produces no intermediates. As shown in the mechanism drawing, the three double bonds in the two starting materials are used to make two new carbon-carbon single bonds and one new carbon-carbon double bond in the product.
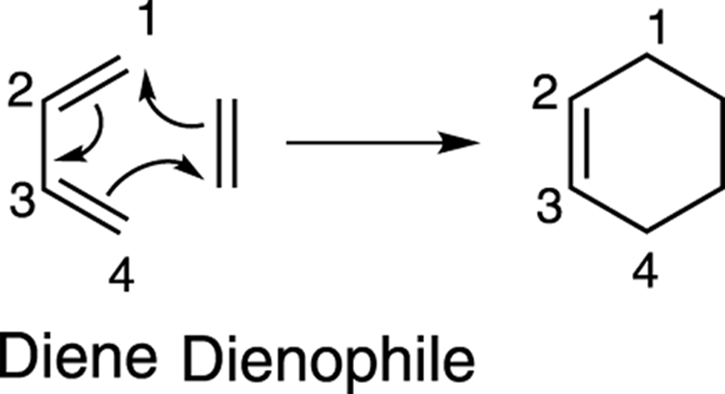
FIGURE 14-5: The mechanism of the Diels–Alder reaction.
Seeing the diene and the dienophile
The Diels–Alder reaction prefers certain dienes and dienophiles. Dienes that are substituted with electron-donating groups react faster than those that are unsubstituted. Conversely, dienophiles react the fastest when they’re substituted with electron-withdrawing groups.
Good electron-donating groups (abbreviated EDG) to put on dienes for the Diels–Alder reaction include ethers (OR), alcohols (OH), and amines (NR2). Good electron-withdrawing groups (abbreviated EWG) for the dienophile include cyano groups (CN), nitro groups (NO2), and all the carbonyl compounds (including esters, aldehydes, acids, ketones, and so on). These groups are shown in Figure 14-6.
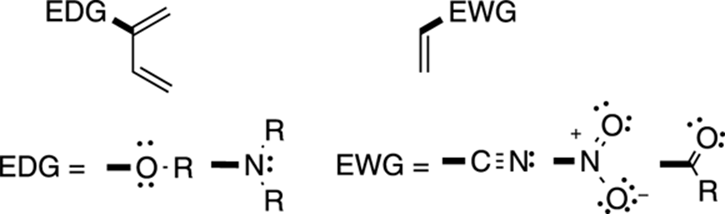
FIGURE 14-6: Some diene and dienophile preferences.
Another requirement of the diene is that it adopt the s-cis conformation so that the two double bonds are in the proper orientation to undergo the reaction. The s-cis conformation is the one in which both of the double bonds are on the same side of the carbon-carbon single bond that separates the two double bonds, as shown in Figure 14-7. Dienes in rings react very quickly in the Diels–Alder reaction because they’re locked in the s-cis conformation.
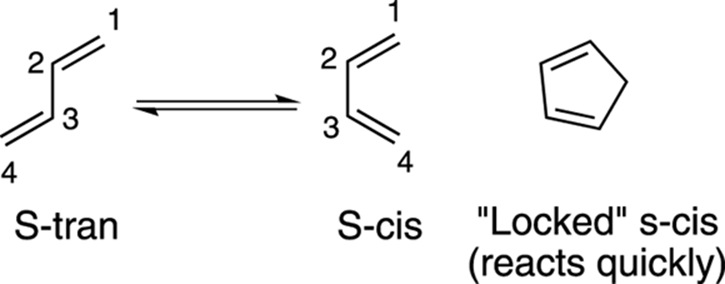
FIGURE 14-7: The s-cis and s-trans conformations.
The stereochemistry of addition
One of the most valuable features of the Diels–Alder reaction is that the reaction is stereoselective — that is, it preferentially forms one stereoisomer over another. (For a discussion of stereochemistry, check out Chapter 6.) With a disubstituted dienophile, if the two substituents on the dienophile start cis, they end up cis in the product; if they start trans, they end up trans in the product, as shown in Figure 14-8.
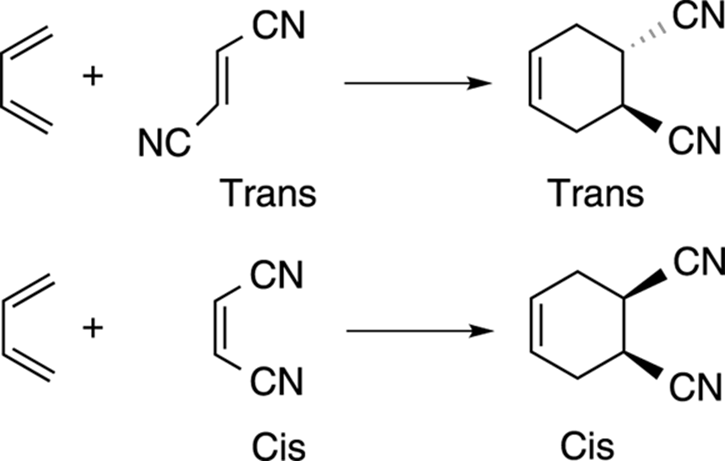
FIGURE 14-8: Stereochemistry of addition in a Diels–Alder reaction.
Seeing bicyclic products
If the diene is in a ring, you get bicyclic products from the Diels–Alder reaction, as shown in Figure 14-9. And if the two substituents on the dienophile are cis, the two stereoisomers could end up going down relative to the carbon bridge in the product, called endo addition, or sticking up toward the carbon bridge, called exo addition. The Diels–Alder reaction preferentially forms the endo product.
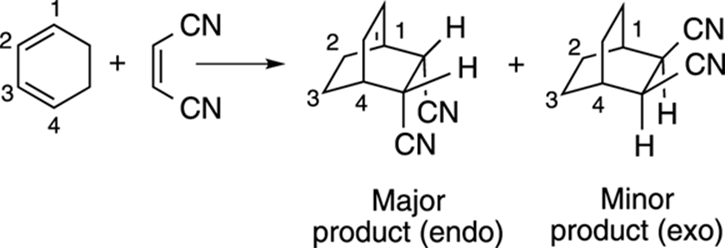
FIGURE 14-9: The endo and exo products of a Diels–Alder reaction.
Problem Solving: Determining Products of Diels–Alder Reactions
Diels–Alder reactions can look confusing, but following these four simple steps will help you to determine the products of these reactions:
1. Orient the diene and the dienophile correctly.
In this step, you make sure that the double bonds are oriented correctly (the diene double bonds are pointing in the direction of the dienophile), and that the diene is in the s-cis conformation (if it isn’t, you need to rotate it so that it is).
2. Number the diene carbons (1 through 4).
You can start the numbering on either end of the diene. The numbering is just a way for you to keep track of where bonds should be formed to make the product.
3. Work the reaction.
Make a bond from the number-one carbon on the diene to one side of the dienophile. Then make a bond from the number-four carbon to the other side of the dienophile. Get rid of the two double bonds in the starting material between carbons 1 and 2 and 3 and 4, and put a double bond between carbon 2 and 3 in the product.
4. Make sure you have the correct stereochemistry.
If you start with a cis disubstituted dienophile, the product should be substituted cis; if the dienophile starts trans, it should end up trans. With bicyclo products, make sure that you show endo stereochemistry in the product.
Now try taking the problem shown in Figure 14-10 through the four steps.
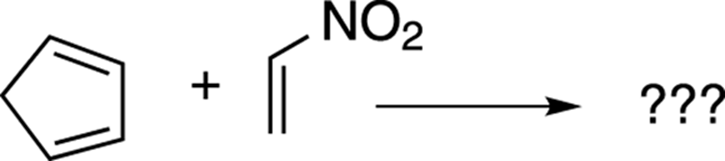
FIGURE 14-10: A Diels–Alder reaction.
Step 1: Orienting the molecule
Start by orienting the diene so that the two double bonds in the diene point in the direction of the dienophile (with the single bond that connects the double bonds oriented away from the dienophile). In this case, you need to rotate the diene 180 degrees, as shown in Figure 14-11. Because this diene is in a ring, you don’t have to worry about twisting the diene to the s-cis conformation because dienes in rings are locked in the s-cis conformation.
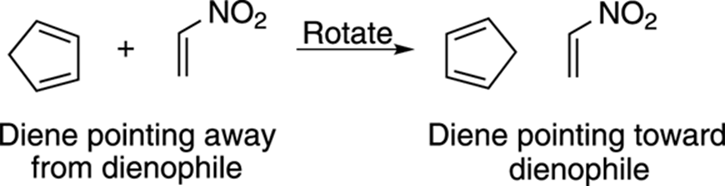
FIGURE 14-11: Orienting the diene.
Step 2: Numbering the diene
Next, number the carbons on the diene (1 through 4). Start the numbering on any side of the diene. In Figure 14-12, the numbering begins on the top.
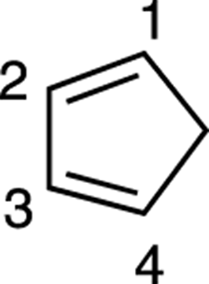
FIGURE 14-12: Diene numbering.
Step 3: Making the bonds
Now that everything’s lined up, take a deep breath and work the reaction. You make bonds between the number-one carbon on the diene and the dienophile, and between the number-four carbon and the dienophile. You end with a double bond between carbons two and three. Notice that in the reaction shown in Figure 14-13, the fifth carbon in the diene ring is flipped up, forming a pointed “bridge” between carbons one and four.

FIGURE 14-13: Two different ways of visualizing this Diels–Alder reaction.
Step 4: Checking the stereochemistry
Make sure you have the correct stereochemistry. Because you have a bicyclic product, you need to make sure that the nitro substituent is endo (points down from the carbon bridge at the top of the molecule). And that’s it!