Process Technology: An Introduction - Haan A.B. 2015
13 Crystallization and precipitation
13.2 Fundamentals
The design of a crystallization process requires information on the solute solubility, metastable limits, nucleation characteristics, and crystal growth. Solubility and phase relationships influence the choice of crystallizer and the method of operation. Although equilibrium and equilibrium calculations are very important, they alone are not sufficient for a complete crystallizer design as for other equilibrium-staged separations. Equilibrium calculations can tell the total mass of crystals produced, but cannot tell the size and the number of crystals produced. To include the crystal size distribution in the design calculations, nucleation and crystal growth rates must be incorporated together with population balances. Nucleation leads to the formation of crystals. Growth is the enlargement of crystals caused by deposition of solid material on and existing surface. The relative rates at which nucleation and growth occur determine the crystal size distribution. Acceptable operating conditions for the minimization of uncontrolled nucleation and encrustation of heat-exchange surfaces are defined by metastable limits.
13.2.1 Solid-liquid equilibria
Accurate solid-liquid equilibrium data are essential to evaluate the process design options for crystallization processes. A saturated solution is a solution which is in thermodynamic equilibrium with the solid phase of its solute at a specified temperature. The saturation solubility of a solid is given by the fundamental thermodynamic relationships for equilibrium between a solid and a liquid phase. When the effect of differences between the heat capacities of the liquid and solid is neglected, the saturation solubility is given by
![]()
(13.1)
where xs is the solid solubility (mole fraction) in the solvent, γs is the liquid phase activity coefficient, ΔHM is the enthalpy of melting, TM(K) is the solid melting temperature, and T(K) is the system temperature. For ideal solutions, eq. (13.1) reduces to the van’t Hoff relationship:
![]()
(13.2)
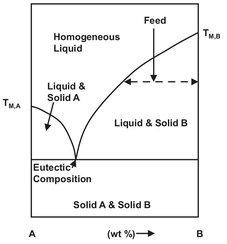
Fig. 13.2: Eutectic solid-liquid phase diagram.
With this equation the solute solubility for an ideal solution can be calculated from the heat of fusion and the pure-solid melting temperature alone. Thus the solubility depends only on the properties of the solute and is independent of the nature of the solvent. A common type of solid-liquid phase diagram is the binary eutectic, shown in Fig. 13.2, in which a pure solid component is formed by cooling an unsaturated solution until solids appear. Continued cooling will increase the yield of pure component. At the eutectic point both components solidify and additional purification is not possible.
13.2.2 Supersaturation and metastability
The kinetic phenomena that influence crystal size distributions are nuclation and growth. The driving force for both these phenomena is supersaturation, defined as the deviation from thermodynamic equilibrium. If a solution or melt is slowly cooled, the temperature will gradually drop until the saturation point is reached. At this point however, no spontaneous solidification will take place, because the rate of nucleation is negligible. Change of phase only occurs when a certain degree of supercooling provides the required driving force to initiate nucleation. This region of supercooling is known as the metastable region where the solution contains more dissolved solute than that given by the equilibrium saturation value. This is illustrated in Fig. 13.3, showing the solubility curve, the supersaturation line of a solution, and the metastable region in which the nucleation rate is very low. The degree of supersaturation can be expressed in many different ways. In crystallization it is common to use the difference between the solute concentration and the concentration at equilibrium:
![]()
(13.3)
where c is the actual solution concentration and c* the equilibrium saturation value. Another common expression is the relative supersaturation s, given by the ratio of the difference between the solute concentration and the equilibrium concentration to the equilibrium concentration:
![]()
(13.4)
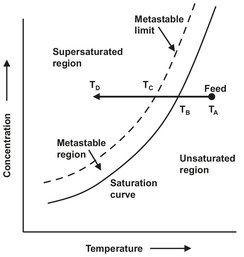
Fig. 13.3: Schematic depiction of supersaturation.
Solution concentration may be expressed in a variety of units. For general mass balance calculations, kilograms anhydrate per kilogram of solvent or kilograms hydrate per kilograms of free solvent are the most convenient. The former avoids complications if different phases can crystallize over the temperature range considered. The importance of supersaturation in setting nucleation and growth rates is clarified in Fig. 13.4, which schematically shows the influence of supersaturation on nucleation and growth. The key aspects in this figure are the qualitative relationships of the two forms of nucleation to growth and to each other. Growth rate and secondary nucleation kinetics are low-order (shown as linear) functions of supersaturation, while primary nucleation follows a high order dependence on supersaturation. Design of a crystallizer to produce a desired crystal size distribution requires the quantification of nucleation and growth rates to externally controlled variables and to supersaturation.
13.2.3 Nucleation
In crystallization, nucleation is the formation of a solid phase from a liquid phase. The process differs from growth in that a new crystal results from the transfer of solute from the liquid to the solid. Because it is the phenomenon of crystal formation, nucleation sets the character of the crystallization process, and it is therefore the most critical component in relating crystallizer design and operation to crystal size distribution. In primary nucleation, existing crystals are not involved in the nucleation process. Homogeneous primary nucleation occurs in the bulk liquid phase in the absence of crystals or any foreign particles. Heterogeneous primary nucleation occurs on a solid surface such as a dust particle or the vessel wall. Secondary nucleation is heterogeneous nucleation induced by existing crystals.
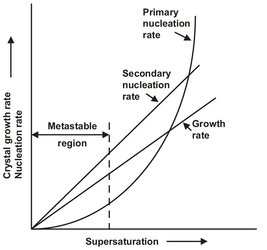
Fig. 13.4: Influence of supersaturation on nucleation and crystal growth rates.
Homogeneous primary nucleation is only observed when the vessel is meticulously cleaned, polished, and closed to avoid the presence of atmospheric dust that will act as heterogeneous nucleation sites. In classical homogeneous nucleation theory a large number of solute units (atoms, molecules, or ions) can aggregate together in a highly supersaturated solution to form a cluster. If the cluster can reach a certain critical size, the free-energy change upon further growth will be negative. Then the cluster is stable and serves as a nucleus for further growth. If the cluster cannot reach the critical size, it redissolves. The simplest expression for the homogeneous primary nucleation rate is
![]()
(13.5)
where k is the Boltzman constant, σ is the surface energy per unit area, ν is molar volume, s is the supersaturation, and A is a constant. The high-order dependence on supersaturation is especially important as a small variation in supersaturation may produce an enormous change in nucleation rate and can lead to production of excessive fines when primary nucleation mechanisms are important.
In heterogeneous primary nucleation the crystal forms around a foreign object such as a dust particle or a crack in the vessel wall. The foreign object lowers the surface energy because the solvent wets the solid. Although eq. (13.5) is applicable, the absolute value of the argument of the exponential decreases by 4 to 5 orders or magnitude. This greatly increases the rate of nucleation at much lower supersaturations, which is of specific importance to industrial systems that are never completely free of suspended solids, less perfectly cleaned, and contain incompletely polished interior surfaces.
Secondary nucleation is the formation of new crystals as a result of the presence of solute crystals through several mechanisms, including initial breeding, contact nucleation, and sheer breeding. In most industrial crystallizers, secondary nucleation is far more important than primary nucleation. This is because continuous and seeded batch crystallizers always contain crystals that can participate in secondary nucleation mechanisms and are operated at low supersaturations to obtain a regular and pure product. The presence of crystals and low supersaturation can only support secondary and not primary nucleation. Initial breeding results from adding seed crystals to a supersaturated solutions to induce nucleation. The presence of growing crystals can also contribute to secondary nucleation by several different mechanisms. Contact nucleation results from nuclei formed by collisions of crystals against each other, against crystallizer internals or with an impellor, agitator or circulation pump. Sheer breeding results when a supersaturated solution flows by a crystal surface and carries along crystal precursors formed in the region of the growing crystal surface.
The ease with which nuclei can be produced by contact nucleation is a clear indication that this mechanism is dominant in many industrial crystallization operations. For design purposes the metastable limit can provide a useful semiempirical approach to correlate the effective or apparent heterogeneous and secondary nucleation rate:
![]()
(13.6)
where c is the solute concentration, cm is the solute concentration at which spontaneous nucleation occurs, and c* is the solute concentration at saturation. The metastable limit must be determined through experimentation. Fortunately cm is very close to c* for many inorganic systems, and satisfactory correlation can be obtained from
![]()
(13.7)
where the parameters kn and n must be evaluated from experimental data. The order for heterogeneous nucleation can range from 2 to 9. For secondary nucleation the order is significantly lower and in the range of 0-3. Many commercial crystallizers operate in the secondary nucleation range.
13.2.4 Crystal growth
Crystal growth is a complex subject, which, like nucleation, is not completely understood and may be expressed in a variety of ways:
· — linear advance rate of an individual crystal face:
· — change in a characteristic dimension of a crystal;
· — rate of change in mass of a crystal.
Adsorption layer or kinetic theories have proven to be quite fruitful in explaining crystal growth. They postulate that there is an “adsorbed” layer of units which is loosely held to the crystal face. These adsorbed units are free to move on the two-dimensional surface, but they have essentially lost one degree of freedom. Thus the unit must crystallize through two-dimensional nucleation. At low supersaturations the energy required for two-dimensional nucleation is considerably less than for normal nucleation. Thus, existing crystals can grow under conditions where three-dimensional nucleation will not occur. Crystal growth will usually occur at supersaturation levels lower than can be explained by the two-dimensional nucleation theory. This happens because growth is much faster at any imperfection where the surface is not a perfect plane. Since crystals are usually not perfect, growth usually proceeds by a “filling-in” process. Small crystals are more likely to be perfect than large crystals and therefore more likely to require two-dimensional nucleation. Large crystals are less likely to be perfect and are more likely to be damaged by the impeller or baffles. Thus, the large crystals can grow by healing kinks, pits, and dislocations. These imperfections are also the reason that even two crystals of the same size may have different growth rates although all conditions appear to be the same. This is called growth rate dispersion. If one of the crystals happens to be more perfect than the other it will have a lower growth rate.
The adsorption theory that explains the crystallization once a unit is at the surface is often incorporated into mass transfer theories that describe the movement of the unit to the surface. In crystallization the picture of the mass transfer process is somewhat different from mass transfer theories for other processes, and different empirical expressions are used. Fig. 13.5 shows a schematic of the mass transfer process that can be broken down into the following seven steps:
· (1) bulk diffusion of solvated units through the film;
· (2) diffusion of solvated units in the adsorbed layer;
· (3) partial or total desolvation;
· (4) surface diffusion of units to growth site;
· (5) incorporation into a lattice;
· (6) counter diffusion of solvent through the adsorbed layer;
· (7) counter diffusion of solvent through the film.
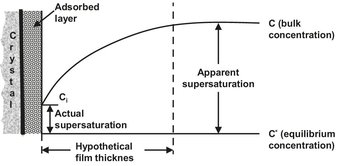
Fig. 13.5: Schematic of crystal growth mass transfer process.
Since it is difficult to include all these steps in a theory, usually a simplified model is used which includes step 1, combines step 2 to 5, and ignores the last two steps. This way a linear growth rate G of the crystal is obtained which is equal in all three dimensions:
![]()
(13.8)
Because of the assumptions involved, it should not be surprising that many systems do not satisfy this law. In these cases a common approach is to introduce an empirical “overall growth-rate order” n which yields for the growth rate a power-law function in which the constants kG and n are determined by fitting experimental crystal growth data:
![]()
(13.9)
Such an approach will only be valid over small ranges of supersaturation. In addition to supersaturation, temperature will also affect the crystal growth rate through diffusion and the crystal rearrangement process. If either mass transfer or surface reaction controls the temperature, the dependence of growth kinetics can often be expressed in terms of an Arrhenius expression:
![]()
(13.10)
When both mechanisms are important this equation is not valid, and Arrhenius plots for crystal growth data tend to give curved instead of straight lines.
13.2.5 Effects of impurities
The presence of impurities can alter the growth rates of crystalline materials significantly. Most common is a decrease in growth rate, which is considered to involve adsorption of the impurity onto the crystal surface. Once located on the surface, the impurity forms a barrier to solute transfer from the solution to the crystal. Another important effect associated with the presence of impurities is that they may change the crystal shape (habit). Habit alteration is considered to result form unequal changes in the growth rates of different crystal faces.