CHEMICAL BIOLOGY
Chemical Strategies for Enzyme Catalysis
Timothy D.H. Bugg, Department of Chemistry, University of Warwick, Coventry, United Kingdom
doi: 10.1002/9780470048672.wecb154
This article will describe the different chemical strategies used by enzymes to achieve rate acceleration in the reactions that they catalyze. The concept of transition state stabilization applies to all types of catalysts. Because enzyme-catalyzed reactions are contained within an active site of a protein, proximity effects caused by the high effective concentrations of reactive groups are important for enzyme-catalyzed reactions, and, depending on how solvent-exposed the active site is, substrate desolvation may be important also. Examples of acid-base catalysis and covalent (nucleophilic) catalysis will be illustrated as well as examples of ''strain'' or substrate destabilization, which is a type of catalysis observed rarely in chemical catalysis. Some more advanced topics then will be mentioned briefly: the stabilization of reactive intermediates in enzyme active sites and the possible involvement of protein dynamics and hydrogen tunneling in enzyme catalysis.
Introduction
Enzymes are biologic catalysts that speed up chemical and biochemical reactions. Three particular characteristics of enzyme catalysis that distinguish enzymes from most man-made catalysts are speed, selectivity, and specificity. Enzymes are capable of rate accelerations of 106-1017 compared with the uncatalyzed reaction. Enzymes are highly selective catalysts; they can recognize and bind a single enantiomer of racemic mixture because the active site of the enzyme is chiral. The specificity of enzyme-catalyzed reactions is very high also; in reactions that generate a new chiral center (e.g., reduction of a ketone to an alcohol), only one enantiomer of the product is obtained. Again, this result is because of the chirality of the enzyme active site and the selective binding of the substrate by the enzyme.
This article will describe the different chemical strategies used by enzymes to achieve rate acceleration in the reactions that they catalyze. The concept of transition state stabilization applies to all types of catalysts. Because enzyme-catalyzed reactions are contained within an active site of a protein, proximity effects caused by the high effective concentrations of reactive groups are important for enzyme-catalyzed reactions, and, depending on how solvent-exposed the active site is, substrate desolvation may be important also. Examples of acid-base catalysis and covalent (nucleophilic) catalysis will be illustrated as well as examples of “strain” or substrate destabilization, which is a type of catalysis observed rarely in chemical catalysis. Some more advanced topics then will be mentioned briefly: the stabilization of reactive intermediates in enzyme active sites and the possible involvement of protein dynamics and hydrogen tunneling in enzyme catalysis.
Transition State Theory for Enzyme Catalysis
The rate of a chemical reaction of substrate S to product P is governed by the activation energy (Eact), which is the difference in free energy between the reagent(s) and the transition state for the reaction. The relationship between rate and Eact is described by the Arrhenius equation:
![]()
The transition state theory for enzyme catalysis predicts that catalysis is achieved by reducing the activation energy for the catalyzed reaction. This reduction in activation energy can be achieved either by stabilization (and hence reduction in free energy) of the transition state by the catalyst or by the catalyst finding some other lower energy pathway for the reaction. To illustrate the above equation, if a catalyst can provide 10 kJ mol-1 of transition stabilization energy for a reaction at 25 °C, then a 55-fold rate acceleration will result, whereas a 20-kJ mol-1 stabilization will give a 3000-fold acceleration and a 40-kJ mol-1 stabilization a 107-fold acceleration!
Figure 1 illustrates the free energy profile of a typical acid-catalyzed chemical reaction that converts a substrate S to a product P. In this case, an intermediate chemical species SH+ is formed on protonation of S. If the activation energy for conversion of SH+ to PH+ is lower than for the conversion of S to P, then the reaction will go faster. It is important at this point to define the difference between an intermediate and a transition state: An intermediate is a stable (or semistable) chemical species formed during the reaction and therefore is a local energy minimum, whereas a transition state, by definition, is a local energy maximum.

Figure 1. Free energy profiles for (a) an acid-catalyzed reaction and (b) an enzyme-catalyzed reaction.
An enzyme-catalyzed reaction can be analyzed thermodynamically in the same way as the acid-catalyzed example, except that enzyme-catalyzed reactions are multistep sequences that involve several intermediates. An enzyme-substrate intermediate ES is formed during binding of the substrate, which then is converted to the enzyme-product complex EP either directly or via one or more additional intermediates.
Under saturating substrate concentrations, the rate of the enzyme-catalyzed reaction will be governed by the activation energy for the conversion of the ES complex to the EP complex. It is clear that if the substrate is bound more tightly by the enzyme, then the size of this activation energy barrier will increase, which leads to a reduced rate. Therefore, for optimum rates of catalysis, enzymes should bind the substrate fairly weakly, but they should selectively bind the transition state of the reaction.
Exactly how do enzymes achieve transition state stabilization? I will illustrate three examples; the first two involve hydrogen-bonding interactions, and the third involves electrostatic interactions. The first example is the well-studied serine protease enzyme, a-chymotrypsin, which cleaves polypeptide substrates via hydrolysis of the peptide bond adjacent to phenylalanine or tyrosine residues. The active site of α-chymotrypsin contains a catalytic triad comprising Ser-195, His-57, and Asp-102. Active site base His-57 deprotonates the hydroxyl side chain of Ser-195, which attacks the amide carbonyl of the substrate, as shown in Fig. 2a (1). The oxyanion intermediate that is generated is stabilized via hydrogen bonding to two backbone amide N-H groups, those of Ser-195 and Gly-193, in the “oxyanion hole,” which is illustrated in Fig. 2b. These hydrogen bonds are formed only with the oxyanion, not with the bound substrate, and hence provide transition state stabilization.
The second example is a sialyltransferase enzyme from Campylobacter jejuni, which catalyzes the transfer of a 9-carbon sialic acid sugar from CMPNeu5Ac to an acceptor substrate (2). Analysis of the crystal structure of this enzyme revealed that the phosphate-leaving group of CMP interacted with two active site tyrosine residues, Tyr-156 and Tyr-162. Replacement of each tyrosine residue by phenylalanine gave 100-fold and 300-fold reduction in kcat, respectively, which indicates that these residues are important in catalysis. As C-O cleavage takes place, the lengthening of the C-O bond and the additional negative charge on the phosphate group lead to much stronger hydrogen bonds with Tyr-156 and Tyr-162 in the transition state, as shown in Fig. 3, and hence transition state stabilization.
The third example is a phosphoryl transfer enzyme, alkaline phosphatase. The active site of alkaline phosphatase contains two Zn2+ ions, with a separation of 3.9 A. One zinc center is used to bind the phosphate monoester substrate, the other to activate Ser-102 for nucleophilic attack on the phosphate group of the substrate via an associative mechanism, as shown in Fig. 4 (3). Situated between the Zn2+ ions is the guanidinium side chain of Arg-166, which provides electrostatic stabilization for the additional negative charge in the transition state of this phospho-transfer reaction.

Figure 2. (a) Mechanism of TS stabilization in a-chymotrypsin. (b) Structure of oxyanion hole in active site.
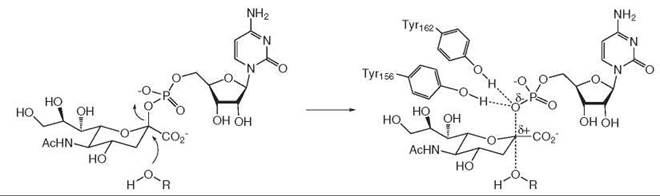
Figure 3. Sialyltransferase.
Proximity Effects
Intramolecular reactions in organic chemistry generally proceed much more rapidly and under much milder reaction conditions than intermolecular reactions because the two reactive groups are “in close proximity” to one another. This effect also operates in enzyme-catalyzed reactions because of the binding of substrate(s) close to the catalytic groups at the enzyme active site.
One example is illustrated in Fig. 5. Acid-catalyzed hydrolysis of sugar glycosides occurs via initial protonation of the departing hydroxyl group. An intramolecular example of glycoside hydrolysis is shown, in which a carboxylic acid group is positioned close to the departing hydroxyl group. The close proximity of the carboxylic group increases the local concentration of H+, which increases the probability of the desired reaction and leads to a 104-fold increase in rate of glycoside hydrolysis. In the enzyme P-galactosidase, the substrate is bound in close proximity to a catalytic glutamic acid residue, which protonates the leaving group. The turnover number (kcat) of the enzyme-catalyzed reaction is 103-fold higher than the intramolecular reaction, which indicates that the enzyme can employ additional strategies to achieve rate acceleration.
Another way of bringing reactants into close proximity, which is encountered commonly in transition metal chemistry, is through metal ion complexation. The coordination of a reactant to a metal ion complex often activates its reactivity and can bring the reactant into close proximity with a second reactant or with a catalytic group. One example, shown in Fig. 6, is a zinc (II) complex of 1,5,9-triazacyclononane, as a model for the enzyme carbonic anhydrase, which contains a zinc (II) cofactor in its active site (4). In the aqua complex, the bound water molecule has a dramatically reduced pKavalue of 7.3, which is similar to the pKa of the active site nucleophilic water. The corresponding cobalt (III) complex catalyzed ester hydrolysis at twice the rate because Co(III) can coordinate both the hydroxide nucleophile and the ester carbonyl via a 5-coordinate intermediate, which is shown in Fig. 6. Many enzymes use metal ion cofactors, which can bind cosubstrates at a metal ion center, close to active site catalytic residues.
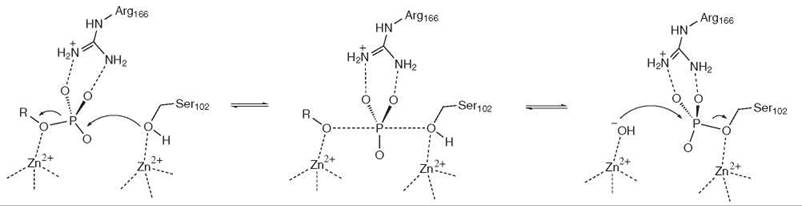
Figure 4. Alkaline phosphatase.

Figure 5. Rate accelerations in glycoside hydrolysis.
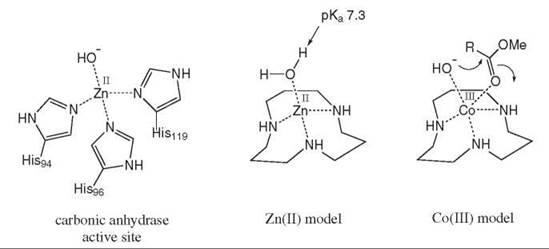
Figure 6. Zinc (II) and cobalt (III) complexes of 1,5,9-triazacyclododecane as mimics of carbonic anhydrase.
Acid/Base Catalysis In Enzymatic Reactions
All enzyme-catalyzed reactions that involve proton transfer use acid or base catalysis, so most enzyme active sites contain acidic or basic amino acid side chains that participate in catalysis. Because enzyme-catalyzed reactions take place close to pH 7, only fairly weak acids and bases are available, as shown in Fig. 7.
Amino acid side chains with pKa values below 7, such as aspartic acid or glutamic acid, will be deprotonated at pH 7 and, therefore, will be used normally in general base catalysis. Amino acid side chains with pKa values above 7, such as lysine or tyrosine, will be protonated at pH 7 and, therefore, will be used normally in general acid catalysis. The imidazole side chain of histidine has a pKa value of 6-8 and, therefore, might be either protonated or deprotonated (or a proportion of each) at pH 7; thus, histidine can be employed as either acid or base in enzyme catalysis and is found widely as an acid/base group in enzymes.
The actual pKa value of an active site catalytic group will be influenced by the particular microenvironment of the active site, which could raise or lower the pKa. For example, the enzyme acetoacetate decarboxylase contains an active site lysine residue that forms an imine link with its substrate; its pKa value was found to be 5.9, which is much less than the expected value of 9. Adjacent to this residue in the active site is a second lysine residue, which in protonated form destabilizes the protonated amine and, therefore, reduces the pKa. Conversely, aspartic acid or glutamic acid residues that are positioned in hydrophobic active sites can have increased pKa values near 7 because the anionic form of the side chain is destabilized.
Enzyme active sites frequently have a pair of acid/base groups, one of which deprotonates one part of the substrate while the other protonates another part of the molecule; this dual action is known as bifunctional catalysis. One example is the enzyme ketosteroid isomerase, whose active site contains two catalytic residues; aspartate-38 acts as a catalytic base, and tyrosine-14 acts as an acidic group (5). The mechanism involves the formation of a dienol intermediate via a concerted step that involves simultaneous deprotonation of the substrate by Asp-14 and protonation of the substrate carbonyl by Tyr-14, as shown in Fig. 8.
Finally, enzymes that bind metal cofactors such as Zn2+ and Mg2+ can use their properties as Lewis acids, for example, electron pair acceptors. An example is the enzyme thermolysin, whose mechanism is illustrated in Fig. 9. In this enzyme, glutamate-143 acts as an active site base to deprotonate water for attack on the amide carbonyl, which is at the same time polarized by coordination by an active site Zn2+ ion (6). The protonated glutamic acid then probably acts as an acidic group for the protonation of the departing amine.

Figure 7. Amino acid side chains involved in acid/base chemistry.

Figure 8. Ketosteroid isomerase mechanism.
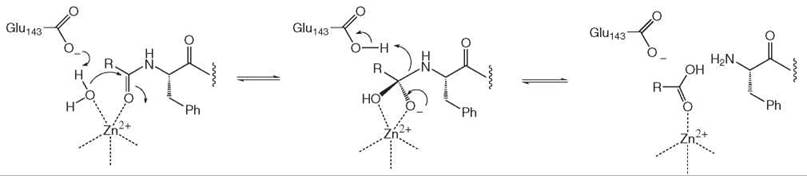
Figure 9. Thermolysin mechanism.
Nucleophilic Catalysis in Enzymatic Reactions
Several enzyme-catalyzed reactions involve the nucleophilic attack of an active site amino acid side chain on the substrate, which results in an immediate reaction that is attached covalently to the enzyme. This type of catalysis is known as nucleophic (or covalent) catalysis.
Several different amino acid side chains can act as nucleophiles in enzyme catalysis. The most powerful nucleophile is the thiol side chain of cysteine, which can be deprotonated to form the even more nucleophilic thiolate anion. One example in which cysteine is used as a nucleophile is the enzyme glyceraldehyde 3-phosphate dehydrogenase, which uses the redox coenzyme NAD+. As shown in Fig. 10, the aldehyde substrate is attacked by an active site cysteine, Cys-149, to form a hemi-thioketal intermediate, which transfers hydride to NAD+ to form an oxidized thioester intermediate (7). Attack of phosphate anion generates an energy-rich intermediate 3-phosphoglycerate.
The ε-amino group of lysine is used in several enzymes to form imine links with ketone groups; for example, it is used in acetoacetate decarboxylase, shown in Fig. 11.
Treatment of this enzyme with substrate and sodium borohydride leads to irreversible enzyme inactivation via in situ reduction of the enzyme-bound imine intermediate by borohydride, which indicates that a covalent link is formed. As mentioned, the pKa of this lysine group is abnormally low at 5.9, which is sufficiently low for it to act as a nucleophile at pH 7 (8).
The other nitrogen nucleophile available to enzymes is the versatile imidazole ring of histidine. This group is used more often for acid/base chemistry, but it is used occasionally as a nucleophile in, for example, phosphotransfer reactions. The serine proteases, such as a-chymotrypsin, which is illustrated in Figs. 2a and (2)b, are classic examples of the participation of serine as a nucleophile. Additional examples exist of nucleophilic mechanisms that employ the hydroxyl groups of threonine and tyrosine and the carboxylate groups of aspartate and glutamate.

Figure 10. Mechanism of B. stearothermophilus GAP DHase.

Figure 11. Acetoacetate decarboxylase mechanism.
Substrate Desolvation
Enzyme active sites sometimes are hydrophobic, buried sites that are excluded largely from water molecules. In these cases, the substrate(s) and enzyme catalytic groups are likely to be “desolvated.” In aqueous solution, a charged nucleophile is surrounded by several layers of water molecules, which greatly reduces its polarity and reactivity. However, a desolvated nucleophile at a water-excluded active site will be a much more potent nucleophile than its counterpart in solution. This effect particularly benefits nucleophilic substitution reactions, found in nucleophilic catalysis.
One recently discovered example is a “fluorinase” enzyme that is involved in the biosynthesis of fluoroacetic acid in Streptomyces cattleya. This enzyme catalyzes the nucleophilic displacement of S-adenosylmethionine by fluoride anion to form 5'-fluoro-5'-deoxy-adenosine (9). Fluoride is a very poor nucleophile in aqueous solution because it is heavily solvated. However, the enzyme can bind fluoride in a desolvated pocket, hydrogen-bonded to the amide backbone N-H and side chain O-H of Ser-158 (see Fig. 12), which increases its reactivity dramatically.
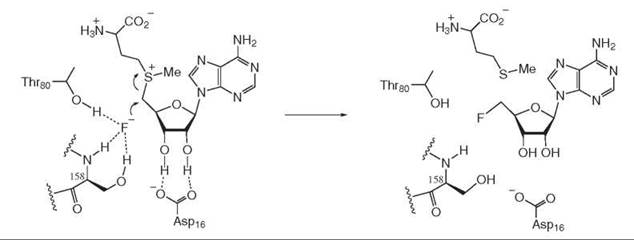
Figure 12. Fluorinase mechanism.
The Use of Strain Energy in Enzyme Catalysis
It is thought that in a small number of enzyme-catalyzed reactions, the enzyme binds the substrate in a strained conformation, which is closer to the transition state than the ground state conformation. In these cases, the difference in energy between the bound conformation and the transition state will be reduced and the reaction will be accelerated (Fig. 13).
One example of this is the enzyme lysozyme, which catalyzes the hydrolysis of the polysaccharide chain of the peptidoglycan layer of bacterial cell walls and, therefore, protects organs such as the human eye from bacterial infection. When the X-ray crystal structure of this enzyme was solved, attempts to model the substrate into the active site were possible only if a “kink” in the substrate was made at the point at which hydrolysis took place (10). The consequence of this kink was a flattening of the sugar ring that was cleaved; this flattened conformation is closer in structure to the oxonium ion formed during C-O bond cleavage, as shown in Fig. 14. Energetically, the enzyme compensated for the local flattening of this ring by more favorable binding interactions elsewhere in the substrate so that substrate binding still was a favorable process, but the activation energy for the bound substrate was reduced by this effect.
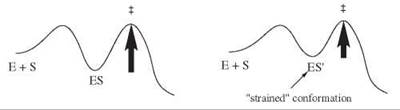
Figure 13. Energy profiles for catalysis by strain.
Stabilization of Reactive Intermediates
One remarkable feature of enzyme-catalyzed reactions is the ability to generate at enzyme sites reactive intermediates, such as carbanions, carbocations, and radicals, that normally would require strong reaction conditions to generate in chemical reactions and would be very unstable in aqueous solution.
Terpene cyclase enzymes catalyze the cyclization of allylic pyrophosphate substrates to form carbocyclic products via carbocation reaction intermediates. One well-studied example is pentalenene synthase (11, 12), which catalyzes the cyclization of farnesyl pyrophosphate to give pentalenene, whose reaction mechanism is shown in Fig. 15. Cyclization of farnesyl pyrophosphate is proposed to form an 11-membered intermediate, humulene, which is followed by a five-membered ring closure to form a bicyclic tertiary carbocation. 1,2-Hydride migration followed by an additional five-membered ring closure gives a tricyclic carbocation, which gives pentalenene, at elimination.
How do these cyclase enzymes control the precise regiochemistry and stereochemistry of these multistep cyclizations? The active site of pentalenene synthase consists of a hydrophobic cleft, which is lined with aromatic and nonpolar residues. It is thought that the carbocation intermediates might be stabilized by the formation of π-cation interactions, with aromatic residues such as phenylalanine, tyrosine, and tryptophan. In pentalenene synthase, replacement of Phe-76 or Phe-77 by Ala gave > 10-fold reduction in activity, which suggests that they may stabilize carbocationic intermediates through π-cation interactions.
One example of an enzyme-catalyzed reaction involving a radical intermediate is the enzyme ribonucleotide reductase, which catalyzes the conversion of ribonucleotides (used for RNA biosynthesis) to 2'-deoxyribonucleotides (used for DNA biosynthesis), as illustrated in Fig. 16. Spectroscopic studies of the R2 subunit of Escherichia coli ribonucleotide reductase have shown that it can form a stable, long-lived, tyrosyl radical species—the first protein radical to be discovered (13).
Single electron transfers within the protein lead to the formation of a cysteine radical on Cys-439 in the enzyme active site, which abstracts, then, the C-3' hydrogen to initiate a radical mechanism, as shown in Fig. 17. Protonation of the C-2' hydroxyl group by Cys-462 and 1-electron transfer from C-3' to C-2' generate a radical at C-2'. Abstraction of a hydrogen atom from a second cysteine, Cys-225, gives the reduced center at C-2' and a Cys-Cys disulfide radical anion, which then transfers 1-electron to C-3' to generate an additional radical at C-3'. Abstraction of a hydrogen atom from Cys-439 regenerates the cysteinyl radical.
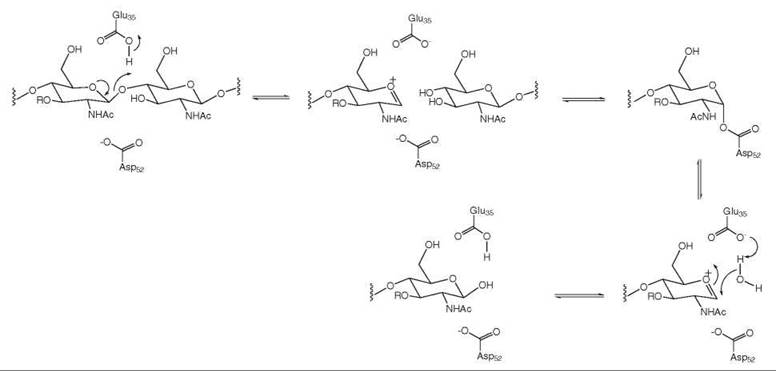
Figure 14. Lysozyme mechanism.
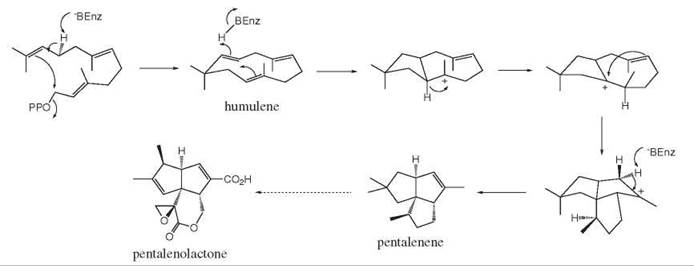
Figure 15. Mechanism of pentalenene synthase.
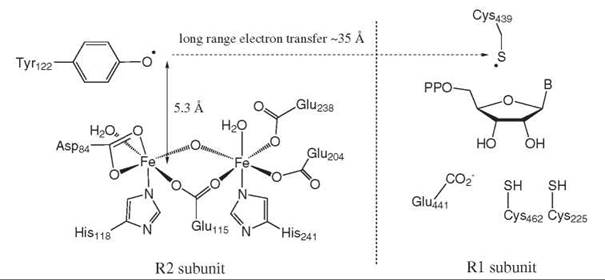
Figure 16. Ribonucleotide reductase reaction; structure of tyrosyl radical.

Figure 17. Mechanism for E. coli ribonucleotide reductase.
The Involvement of Protein Dynamics in Enzyme Catalysis
Examination of protein structure in solution by nuclear magnetic resonance spectroscopy has revealed that a significant amount of internal motion exists in a protein on a timescale of 1 to 10 ns. Such internal motion could transmit kinetic energy from a distant part of the protein to the active site to assist in catalysis. It has been proposed that dynamic fluctations in the protein structure are used by enzymes to organize the enzyme-substrate complex into a reactive conformation.
One example is in the enzyme dihydrofolate reductase, where replacement of Gly-120 to valine disrupts the internal motion of a protein loop, as illustrated in Fig. 18, which leads to a 500-fold reduction in rate; this reduction in rate implies that internal motion is involved in catalysis (14).
In conclusion, enzymes use a variety of strategies to achieve high rates of catalysis. Transition state stabilization seems to be the dominant factor in catalysis, but in some enzymes, the more sophisticated strategies such as substrate destabilization and protein dynamics seem to play an important role.

Figure 18. Illustration of Gly-120 in dihydrofolate reductase.
References
1. Kraut J. α-chymotrypsin. Annu. Rev. Biochem. 1977; 46:331-358.
2. Chiu CPC, Watts AG, Lairson LL, Gilbert M, Lim D, Wakarchuk WW, Withers SG, Strynadka NCJ. Sialyltransferase from Campylobacter jejuni. Nature Str. Biol., 2004; 11:163-170.
3. Holtz KM, Stec B, Kantrowitz ER. Alkaline phosphatase. J. Biol. Chem. 1999; 274:8351-8354.
4. Kimura E, Shiota T, Koike T, Shiro M, Kodama M. Zinc (II) model of carbonic anhydrase. J. Am. Chem. Soc. 1990; 112:5805-5811.
5. Kuliopulos A, Mildvan AS, Shortle D, Talalay P. Ketosteroid isomerase. Biochemistry 1989; 28:149-159.
6. Matthews BW. Thermolysin. Acc. Chem. Res. 1988; 21:333-340.
7. Didierjean C, Corbier C, Fatih M, Favier F, Boschi-Muller S, Branlant G, Aubry A. Glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 2003; 278:12968-12976.
8. Zerner B, Cotts SM, Lederer F, Waters HH, Westheimer FH. Acetoacetate decarboxylase. Biochemistry 1966; 5:813-819.
9. Dong C, Huang FL, Deng H, Schaffrath C, Spencer JB, O’Hagan D, Naismith JH. Fluorinase from Streptomyces cattleya. Nature 2004; 427:561-565.
10. Blake CCF, Johnson LN, Mair GA, North ACT, Phillips DC, Sharma VR. Lysozyme. Proc. Roy. Soc. Ser. B 1967; 167:378-381.
11. Lesburg CA, Zhai G, Cane DE, Christianson DW. Crystal structure of pentalenene synthase: mechanistic insights on terpenoid cyclization reactions in biology. Science 1997; 277:1820-1824.
12. Seemann M, Zhai G, de Kraker JW, Paschall CM, Christianson DW, Cane DE. Pentalenene synthase. Analysis of active site residues by site-directed mutagenesis. J. Am. Chem. Soc. 2002; 124:7681-7689.
13. Stubbe J, van der Donk WA. Ribonucleotide reductase. Chem. Rev. 1998; 98:705-762.
14. Miller GP, Benkovic SJ. Dihydrofolate reductase. Chem. Biol. 1998; 5:R105-113.